PopGeneticsVis
PopGeneticsVis: a toolkit for whole-genome resequencing and bisulfite sequencing downstream visualization in population genetics.
- PopGeneticsVis
- 1. Installation
- 2. Documents
- 2.1 WGRS PLINK PCA Vis
- 2.2 WGRS VCFtools Tajima’D Vis
- 2.3 WGRS PLINK Heterozyg Vis
- 2.4 WGRS VCFtools FST and XPCLR XPCLR Vis
- 2.5 WGBS MethylDackel Cytosine Ratio Vis
- 2.6 MethylKit DMR Analysis and Vis
- 2.6.1 Terminal Running
- 2.6.2 MethylDackel Cytosine Report
./PopGeneticsVis/data/cytosine_report/JJ_G2_1.cytosine_report.txt - 2.6.3 MethylKit DMR results
./PopGeneticsVis/data/cytosine_report/JJ_G2_vs_HN_G2_DMR_CpG_2k.txt - 2.6.4 MethylKit DMR results
./PopGeneticsVis/data/cytosine_report/JJ_G2_vs_HN_G2_DMR_CpG_2k_Sig_Anno.txt
- 2.7 MethylKit Context Vis
- 2.8 MethylKit DMR Violin Vis
PopGeneticsVis
PopGeneticsVis: a toolkit for whole-genome resequencing and bisulfite sequencing downstream visualization in population genetics.
PopGeneticsVis is a comprehensive toolkit specifically designed for the downstream visualization of whole - genome resequencing and bisulfite sequencing in the context of population genetics. It serves as an essential resource for researchers delving into population - level genetic analysis, enabling them to effectively visualize and interpret the complex data generated from these sequencing techniques. This toolkit provides a set of tools and functions that streamline the process of transforming raw sequencing data into meaningful visual representations, facilitating a deeper understanding of genetic patterns, variations, and epigenetic modifications within populations.
1. Installation
1.1 R with Packages
# 1. R: >= v4.3.0
# 2. Packages
# 2.1 CRAN
cran_packages <- c("argparse", "CMplot", "dplyr", "tidyr", "stringr", "data.table", "ggplot2", "ggpubr")
install.packages(cran_packages)
# 2.2 Bioconductor
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
bioconductor_packages <- c("methylKit", "GenomicFeatures", "rtracklayer", "GenomicRanges")
BiocManager::install(bioconductor_packages)
1.2 Download from GitHub Release
https://github.com/benben-miao/PopGeneticsVis/releases
1.3 Clone Git Repository
# 1. Clone
git clone https://github.com/benben-miao/PopGeneticsVis.git
# 2. Test
Rscript ./PopGeneticsVis/bin/plink_pca_vis.R --help
usage: ./PopGeneticsVis/bin/plink_pca_vis.R [-h] --eigenvec EIGENVEC --eigenval EIGENVAL
--sample_pop SAMPLE_POP --select_pops SELECT_POPS
[--x_pc X_PC] [--y_pc Y_PC]
[--point_size POINT_SIZE]
[--point_alpha POINT_ALPHA]
PCA Visualization for PLINK PCA Eigenvec
optional arguments:
-h, --help show this help message and exit
--eigenvec EIGENVEC PLINK PCA results [pca.eigenvec].
--eigenval EIGENVAL PLINK PCA results [pca.eigenval].
--sample_pop SAMPLE_POP
Table with samples in col1, pops in col2
[sample_pop.txt].
--select_pops SELECT_POPS
Select populations ['GroupA,GroupB'].
--x_pc X_PC Which PC for X Axis [1].
--y_pc Y_PC Which PC for Y Axis [2].
--point_size POINT_SIZE
Points size [4].
--point_alpha POINT_ALPHA
Points alpha [0.8].
2. Documents
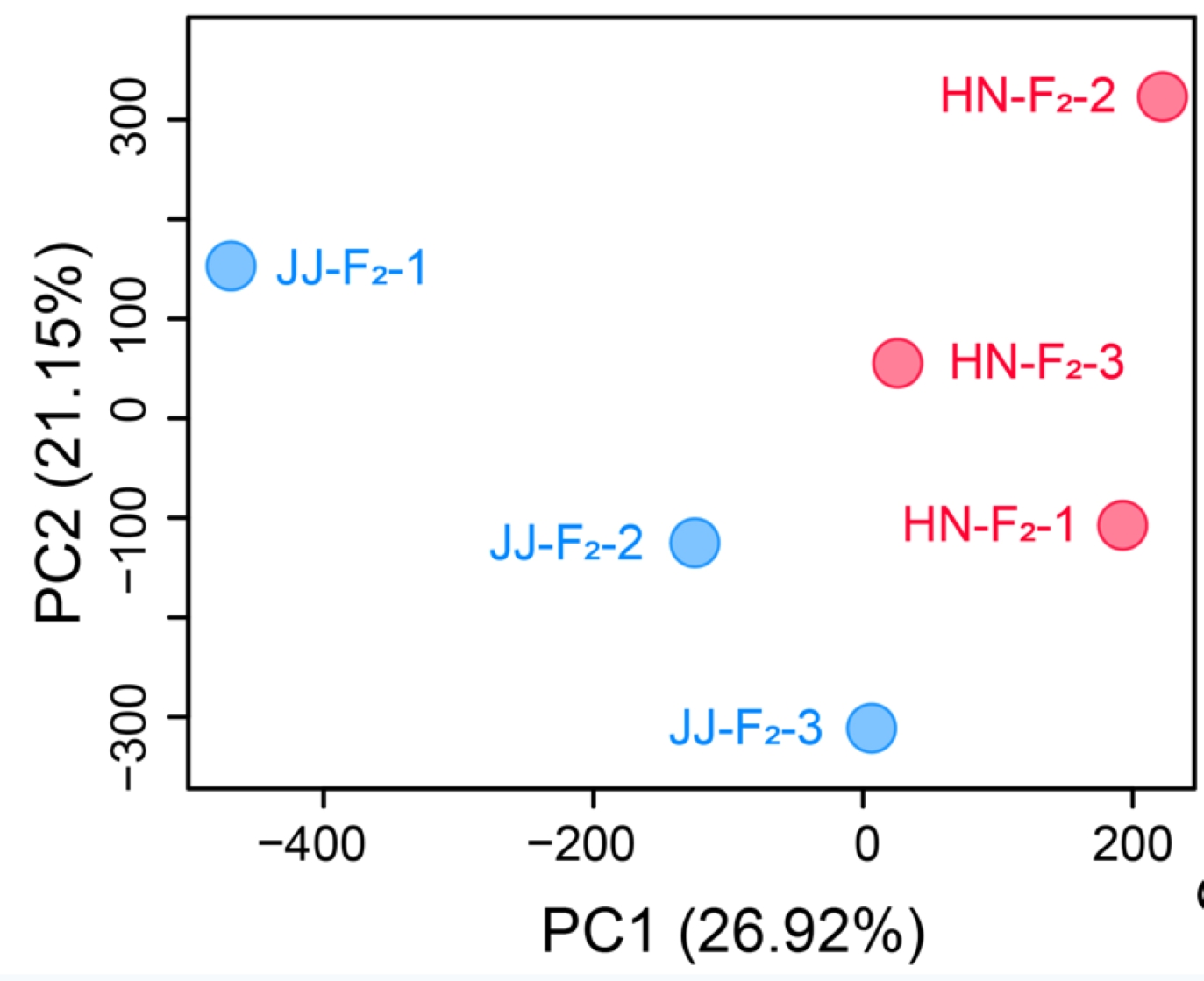
2.1 WGRS PLINK PCA Vis
PLINK (short for Whole-Genome Association Analysis Toolset) is one of the most commonly used tools in genome-wide association studies (GWAS) and population genetics research. Its built-in Principal Component Analysis (PCA) function can efficiently process large-scale genotype data, which is used to correct for population stratification or reveal the genetic structure of samples.
Rscript ./PopGeneticsVis/bin/plink_pca_vis.R --help
usage: ./PopGeneticsVis/bin/plink_pca_vis.R [-h] --eigenvec EIGENVEC --eigenval EIGENVAL
--sample_pop SAMPLE_POP --select_pops SELECT_POPS
[--x_pc X_PC] [--y_pc Y_PC]
[--point_size POINT_SIZE]
[--point_alpha POINT_ALPHA]
PCA Visualization for PLINK PCA Eigenvec
optional arguments:
-h, --help show this help message and exit
--eigenvec EIGENVEC PLINK PCA results [pca.eigenvec].
--eigenval EIGENVAL PLINK PCA results [pca.eigenval].
--sample_pop SAMPLE_POP
Table with samples in col1, pops in col2
[sample_pop.txt].
--select_pops SELECT_POPS
Select populations ['GroupA,GroupB'].
--x_pc X_PC Which PC for X Axis [1].
--y_pc Y_PC Which PC for Y Axis [2].
--point_size POINT_SIZE
Points size [4].
--point_alpha POINT_ALPHA
Points alpha [0.8].
2.1.1 Termial Running
Rscript \
./PopGeneticsVis/bin/plink_pca_vis.R \
--eigenvec ./PopGeneticsVis/data/plink_pca/pca.eigenvec \
--eigenval ./PopGeneticsVis/data/plink_pca/pca.eigenval \
--sample_pop ./PopGeneticsVis/data/plink_pca/samples_pops.txt \
--select_pops "JJ_G2,HN_G2" \
--x_pc 1 --y_pc 2 \
--point_size 4 --point_alpha 0.8
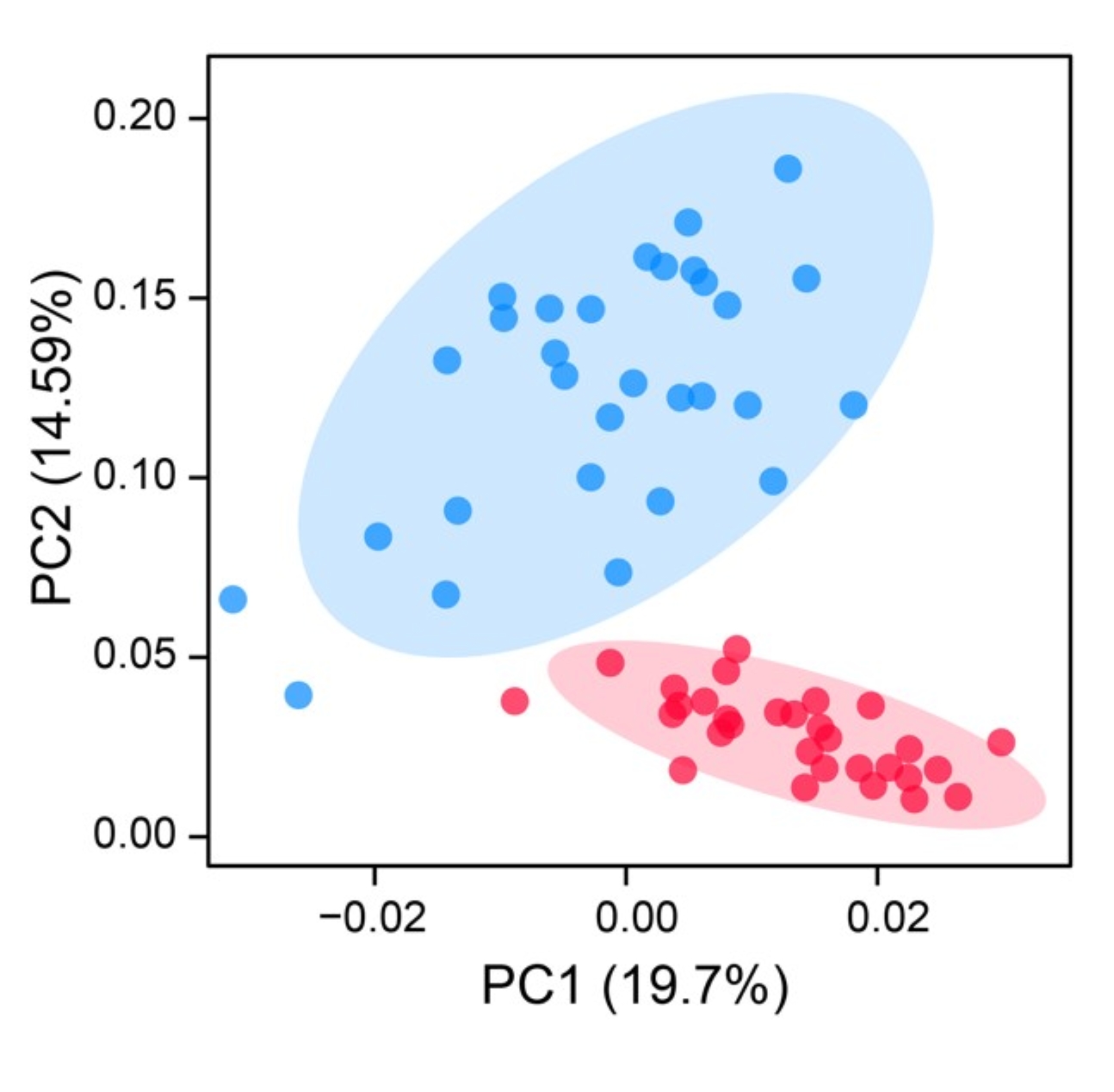
2.1.2 PLINK PCA results ./PopGeneticsVis/data/plink_pca/pca.eigenvec
head -n 10 ./PopGeneticsVis/data/plink_pca/pca.eigenvec
#FID IID PC1 PC2 PC3 PC4 PC5 PC6 PC7 PC8 PC9 PC10
HN_G2_1 HN_G2_1 0.04043 -0.0253789 0.0367277 0.00133413 -0.100252 -0.0905802 0.0113605 -0.0137787 -0.00668236 0.0167026
HN_G2_10 HN_G2_10 0.0212835 0.00859226 -0.0506521 -0.023103 0.0270852 -0.0357936 0.00354596 -0.020354 1.16024e-05 0.0141656
HN_G2_11 HN_G2_11 0.0127448 0.00981976 -0.067746 -0.0214325 -0.0301917 -0.0183634 -0.0200866 -0.00592332 -0.0141271 0.14919
HN_G2_12 HN_G2_12 0.0172671 0.022466 -0.0228558 0.0474678 -0.05247 0.0371412 -0.0215446 -0.0304079 0.0416183 0.0017122
HN_G2_13 HN_G2_13 0.0292439 0.0168136 -0.00497563 0.0267906 -0.0675681 0.0251754 -0.00228862 -0.0429644 0.0618089 -0.0117392
HN_G2_14 HN_G2_14 0.0158522 0.0359309 -0.0488923 0.0203213 0.00240803 -0.012922 0.0210997 -0.0185264 0.0167203 0.0338862
HN_G2_15 HN_G2_15 0.00289866 -0.00861881 -0.0898324 0.0208598 0.0670552 -0.173035 -0.035361 -0.0510791 -0.0171244 -0.0316206
HN_G2_16 HN_G2_16 0.0265133 0.00541896 -0.045698 -0.0219957 0.0469065 0.0547635 0.0421172 -0.0974258 -0.0106709 0.0287779
HN_G2_17 HN_G2_17 0.0250258 0.0229324 -0.0335188 0.00408936 -0.00744732 -0.0204986 0.023825 -0.010836 0.069014 0.0448165
Click to unfold the Table
| #FID | IID | PC1 | PC2 | PC3 | PC4 | PC5 | PC6 | PC7 | PC8 | PC9 | PC10 | |----------|----------|------------|-------------|-------------|------------|-------------|------------|-------------|-------------|-------------|------------| | HN_G2_1 | HN_G2_1 | 0.04043 | -0.0253789 | 0.0367277 | 0.00133413 | -0.100252 | -0.0905802 | 0.0113605 | -0.0137787 | -0.00668236 | 0.0167026 | | HN_G2_10 | HN_G2_10 | 0.0212835 | 0.00859226 | -0.0506521 | -0.023103 | 0.0270852 | -0.0357936 | 0.00354596 | -0.020354 | 1.16E-05 | 0.0141656 | | HN_G2_11 | HN_G2_11 | 0.0127448 | 0.00981976 | -0.067746 | -0.0214325 | -0.0301917 | -0.0183634 | -0.0200866 | -0.00592332 | -0.0141271 | 0.14919 | | HN_G2_12 | HN_G2_12 | 0.0172671 | 0.022466 | -0.0228558 | 0.0474678 | -0.05247 | 0.0371412 | -0.0215446 | -0.0304079 | 0.0416183 | 0.0017122 | | HN_G2_13 | HN_G2_13 | 0.0292439 | 0.0168136 | -0.00497563 | 0.0267906 | -0.0675681 | 0.0251754 | -0.00228862 | -0.0429644 | 0.0618089 | -0.0117392 | | HN_G2_14 | HN_G2_14 | 0.0158522 | 0.0359309 | -0.0488923 | 0.0203213 | 0.00240803 | -0.012922 | 0.0210997 | -0.0185264 | 0.0167203 | 0.0338862 | | HN_G2_15 | HN_G2_15 | 0.00289866 | -0.00861881 | -0.0898324 | 0.0208598 | 0.0670552 | -0.173035 | -0.035361 | -0.0510791 | -0.0171244 | -0.0316206 | | HN_G2_16 | HN_G2_16 | 0.0265133 | 0.00541896 | -0.045698 | -0.0219957 | 0.0469065 | 0.0547635 | 0.0421172 | -0.0974258 | -0.0106709 | 0.0287779 | | HN_G2_17 | HN_G2_17 | 0.0250258 | 0.0229324 | -0.0335188 | 0.00408936 | -0.00744732 | -0.0204986 | 0.023825 | -0.010836 | 0.069014 | 0.0448165 |2.1.3 PLINK PCA results ./PopGeneticsVis/data/plink_pca/pca.eigenval
cat ./PopGeneticsVis/data/plink_pca/pca.eigenval
9.91596
7.34118
5.66223
4.90547
4.37931
4.00635
3.89641
3.54522
3.51559
3.16454
2.1.4 WGRS ./PopGeneticsVis/data/plink_pca/samples_pops.txt
head -n 10 ./PopGeneticsVis/data/plink_pca/samples_pops.txt
HN_G2_1 HN_G2
HN_G2_2 HN_G2
HN_G2_3 HN_G2
HN_G2_4 HN_G2
HN_G2_5 HN_G2
HN_G2_6 HN_G2
HN_G2_7 HN_G2
HN_G2_8 HN_G2
HN_G2_9 HN_G2
HN_G2_10 HN_G2
2.2 WGRS VCFtools Tajima’D Vis
In population genetics research, Tajima’s D test is one of the core methods for evaluating the deviation of DNA sequence polymorphism from the neutral evolution model, and VCFtools (Variant Call Format tools) is a commonly used tool for quickly calculating Tajima’s D values based on VCF files (a standard format for storing genetic variation data).
usage: ./PopGeneticsVis/bin/vcftools_tajimad_vis.R [-h] --tajimad_win TAJIMAD_WIN --select_pops SELECT_POPS
[--box_outliers BOX_OUTLIERS]
VCFtools TajimaD Violin Box Vis
optional arguments:
-h, --help show this help message and exit
--tajimad_win TAJIMAD_WIN
VCFtools TajimaD with ordered POP [all.Tajima.D].
--select_pops SELECT_POPS
Select populations ['GroupA,GroupB'].
--box_outliers BOX_OUTLIERS
Show box outliers uppercase [FALSE].
2.2.1 Terminal Running
Rscript \
./PopGeneticsVis/bin/vcftools_tajimad_vis.R \
--tajimad_win ./PopGeneticsVis/data/vcftools_tajimad/all.Tajima.D \
--select_pops "JJ_G2,HN_G2" \
--box_outliers TRUE
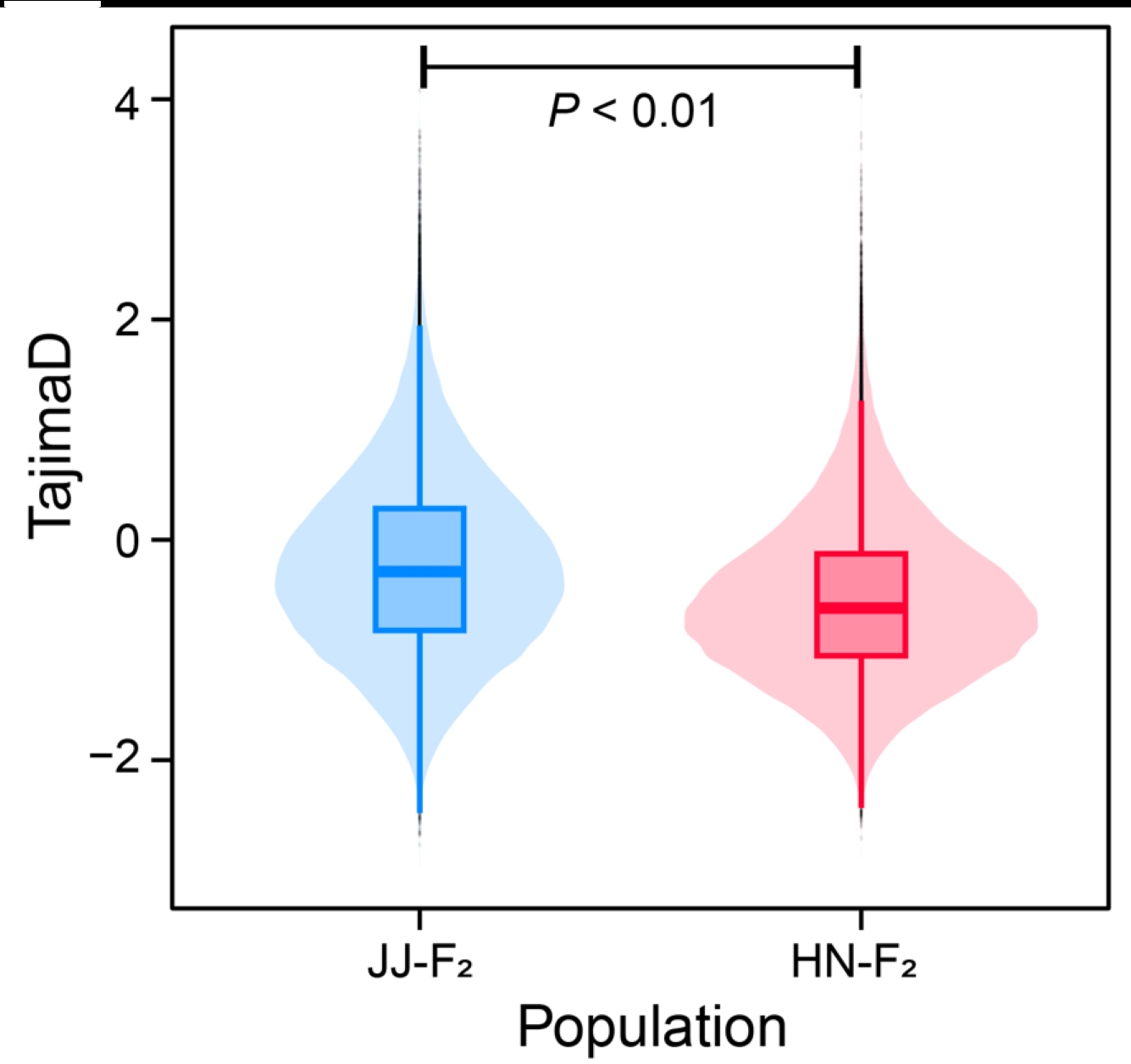
2.2.2 VCFtools Tajima’D results ./PopGeneticsVis/data/vcftools_tajimad/all.Tajima.D
head -n 10 ./PopGeneticsVis/data/vcftools_tajimad/all.Tajima.D
CHROM BIN_START N_SNPS TajimaD POP
11 10000 4 -1.51583 HN_G2
11 20000 0 nan HN_G2
11 30000 3 -0.504447 HN_G2
11 40000 0 nan HN_G2
11 50000 0 nan HN_G2
11 60000 0 nan HN_G2
11 70000 0 nan HN_G2
11 80000 0 nan HN_G2
11 90000 0 nan HN_G2
Click to unfold the Table
| **CHROM** | **BIN_START** | **N_SNPS** | **TajimaD** | **POP** | |-----------|---------------|------------|-------------|---------| | 11 | 10000 | 4 | -1.51583 | HN_G2 | | 11 | 20000 | 0 | nan | HN_G2 | | 11 | 30000 | 3 | -0.504447 | HN_G2 | | 11 | 40000 | 0 | nan | HN_G2 | | 11 | 50000 | 0 | nan | HN_G2 | | 11 | 60000 | 0 | nan | HN_G2 | | 11 | 70000 | 0 | nan | HN_G2 | | 11 | 80000 | 0 | nan | HN_G2 | | 11 | 90000 | 0 | nan | HN_G2 |2.3 WGRS PLINK Heterozyg Vis
In PLINK (a commonly used tool for genome-wide association studies), Heterozyg (Heterozygosity) is a core indicator for measuring the genetic diversity of samples or loci. It reflects the degree to which an individual carries two different alleles (such as A/T) at a specific locus, or the frequency of the distribution of heterozygous alleles at a locus in a population.
usage: ./PopGeneticsVis/bin/plink_heterozyg_vis.R [-h] --het_data HET_DATA --sample_pop SAMPLE_POP --select_pops
SELECT_POPS [--point_size POINT_SIZE]
PLINK Heterozyg Violin Box Vis
optional arguments:
-h, --help show this help message and exit
--het_data HET_DATA PLINK Heterozyg results [het.het].
--sample_pop SAMPLE_POP
Table with IID in col1 and Population in col2
[sample_pop.txt].
--select_pops SELECT_POPS
Select populations ['GroupA,GroupB'].
--point_size POINT_SIZE
Point size [2].
2.3.1 Terminal Running
Rscript \
./PopGeneticsVis/bin/plink_heterozyg_vis.R \
--het_data ./PopGeneticsVis/data/plink_het/het.het \
--sample_pop ./PopGeneticsVis/data/plink_het/samples_pops.txt \
--select_pops "JJ_G2,HN_G2" \
--point_size 2
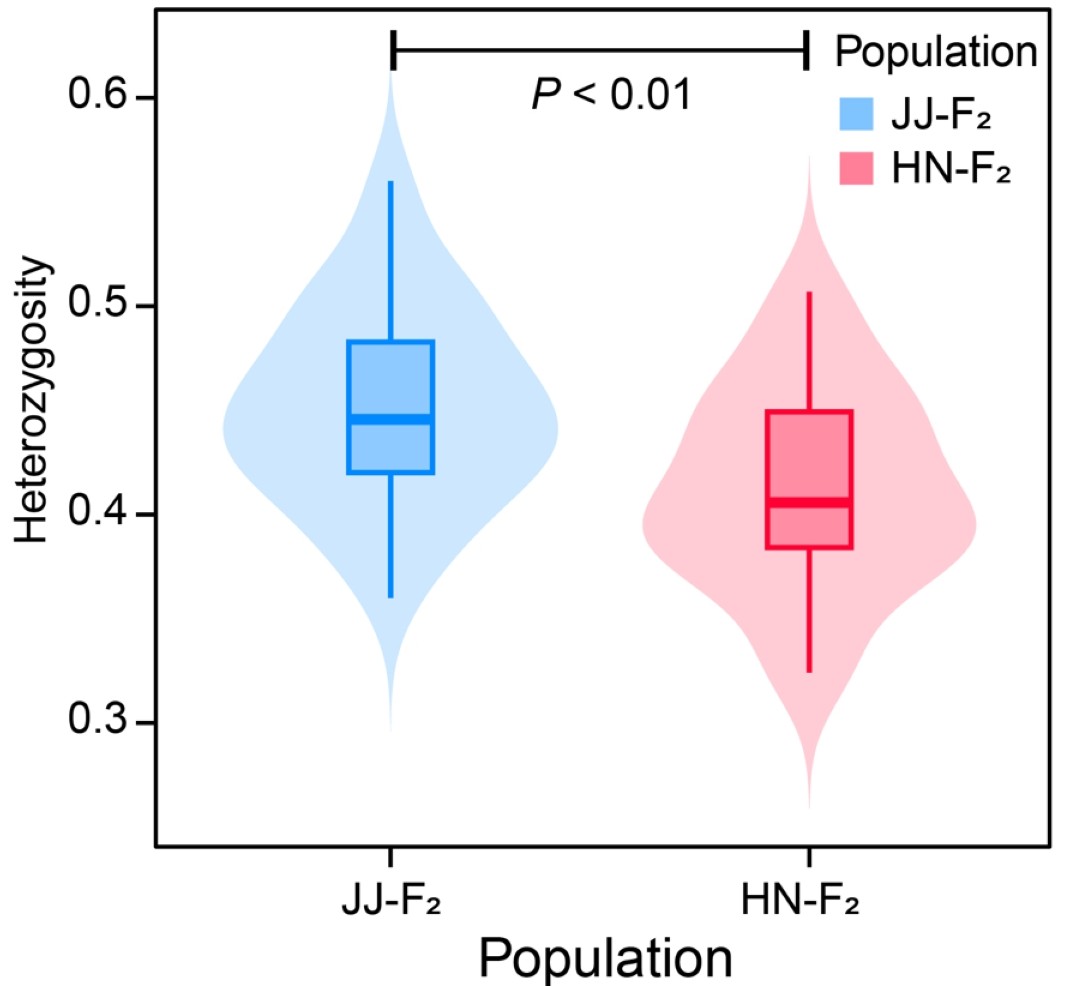
2.3.2 PLINK Heterozyg results ./PopGeneticsVis/data/plink_het/het.het
head -n 10 ./PopGeneticsVis/data/plink_het/het.het
#FID IID O(HOM) E(HOM) OBS_CT F
HN_G2_1 HN_G2_1 19048512 1.81455e+07 20585181 0.370148
HN_G2_10 HN_G2_10 19053719 1.804e+07 20467745 0.417557
HN_G2_11 HN_G2_11 18950164 1.79458e+07 20366876 0.414843
HN_G2_12 HN_G2_12 19064469 1.79607e+07 20383072 0.455651
HN_G2_13 HN_G2_13 19222145 1.8105e+07 20549246 0.457051
HN_G2_14 HN_G2_14 18908338 1.8013e+07 20444064 0.368286
HN_G2_15 HN_G2_15 19256769 1.80381e+07 20474025 0.50029
HN_G2_16 HN_G2_16 19041670 1.80191e+07 20453626 0.420034
HN_G2_17 HN_G2_17 19309606 1.81539e+07 20603448 0.471799
Click to unfold the Table
| **#FID** | **IID** | **O(HOM)** | **E(HOM)** | **OBS_CT** | **F** | |----------|----------|------------|------------|------------|----------| | HN_G2_1 | HN_G2_1 | 19048512 | 1.81E+07 | 20585181 | 0.370148 | | HN_G2_10 | HN_G2_10 | 19053719 | 1.80E+07 | 20467745 | 0.417557 | | HN_G2_11 | HN_G2_11 | 18950164 | 1.79E+07 | 20366876 | 0.414843 | | HN_G2_12 | HN_G2_12 | 19064469 | 1.80E+07 | 20383072 | 0.455651 | | HN_G2_13 | HN_G2_13 | 19222145 | 1.81E+07 | 20549246 | 0.457051 | | HN_G2_14 | HN_G2_14 | 18908338 | 1.80E+07 | 20444064 | 0.368286 | | HN_G2_15 | HN_G2_15 | 19256769 | 1.80E+07 | 20474025 | 0.50029 | | HN_G2_16 | HN_G2_16 | 19041670 | 1.80E+07 | 20453626 | 0.420034 | | HN_G2_17 | HN_G2_17 | 19309606 | 1.82E+07 | 20603448 | 0.471799 |2.3.3 WGRS ./PopGeneticsVis/data/plink_het/samples_pops.txt
head -n 10 ./PopGeneticsVis/data/plink_het/samples_pops.txt
HN_G2_1 HN_G2
HN_G2_2 HN_G2
HN_G2_3 HN_G2
HN_G2_4 HN_G2
HN_G2_5 HN_G2
HN_G2_6 HN_G2
HN_G2_7 HN_G2
HN_G2_8 HN_G2
HN_G2_9 HN_G2
HN_G2_10 HN_G2
2.4 WGRS VCFtools FST and XPCLR XPCLR Vis
VCFtools (Variant Call Format tools) is a core set of tools for processing VCF (Variant Call Format) files in population genetics, while FST (Fixation Index) is a key indicator for measuring the degree of genetic differentiation between populations. The combination of the two can efficiently analyze the genetic differences between different populations at the genomic level, and is widely used in fields such as population structure research, domestication site localization, and adaptive evolution analysis.
usage: ./PopGeneticsVis/bin/cmplot_fst_xpclr.R [-h] [--method {FST,XPCLR}] --fst_xpclr FST_XPCLR
[--plot_type {m,c,d,q}]
CMplot for FST or XPCLR results annotation
optional arguments:
-h, --help show this help message and exit
--method {FST,XPCLR} SNP selection methods [FST].
--fst_xpclr FST_XPCLR
FST or XPCLR results with anno [fst.anno].
--plot_type {m,c,d,q}
Plot type m(Manhattan), c(Circle), d(Density), q(QQ-
plot) [m].
2.4.1 Terminal Running
Rscript \
./PopGeneticsVis/bin/cmplot_fst_xpclr.R \
--method FST \
--fst_xpclr ./PopGeneticsVis/data/vcftools_fst/FST_JJ_G2_vs_HN_G2.windowed.weir.fst.chr.anno \
--plot_type d
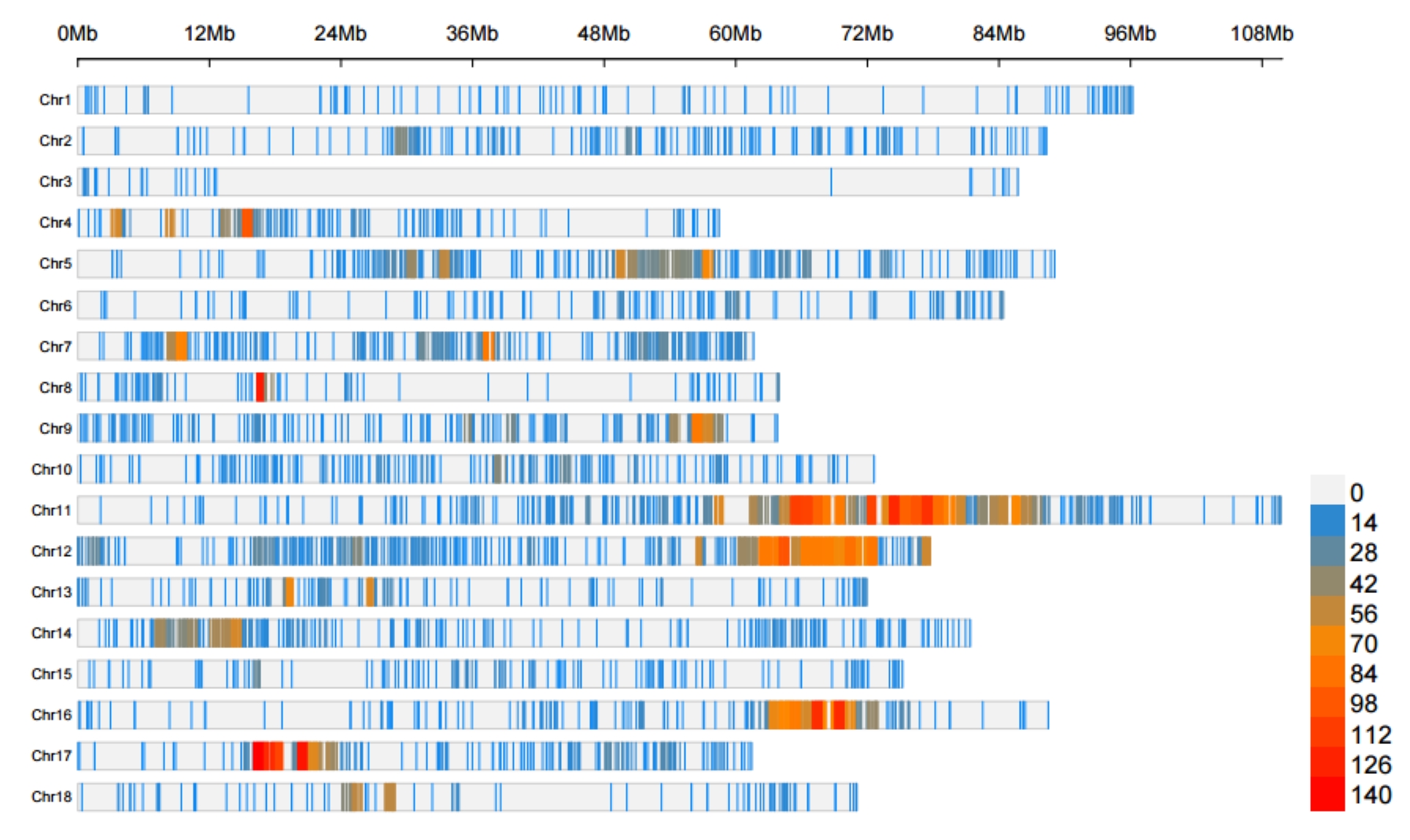
Rscript \
./PopGeneticsVis/bin/cmplot_fst_xpclr.R \
--method FST \
--fst_xpclr ./PopGeneticsVis/data/vcftools_fst/FST_JJ_G2_vs_HN_G2.windowed.weir.fst.chr.anno \
--plot_type m
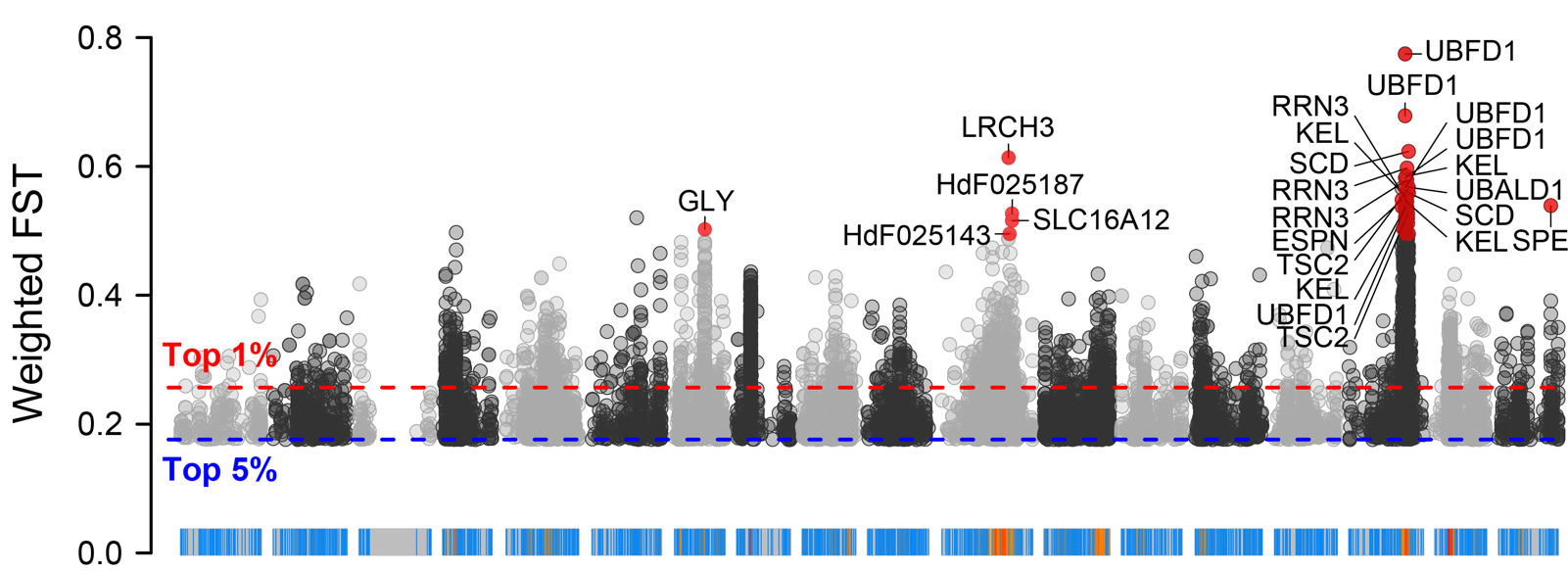
Rscript \
./PopGeneticsVis/bin/cmplot_fst_xpclr.R \
--method FST \
--fst_xpclr ./PopGeneticsVis/data/vcftools_fst/FST_JJ_G2_vs_HN_G2.windowed.weir.fst.chr.anno \
--plot_type c
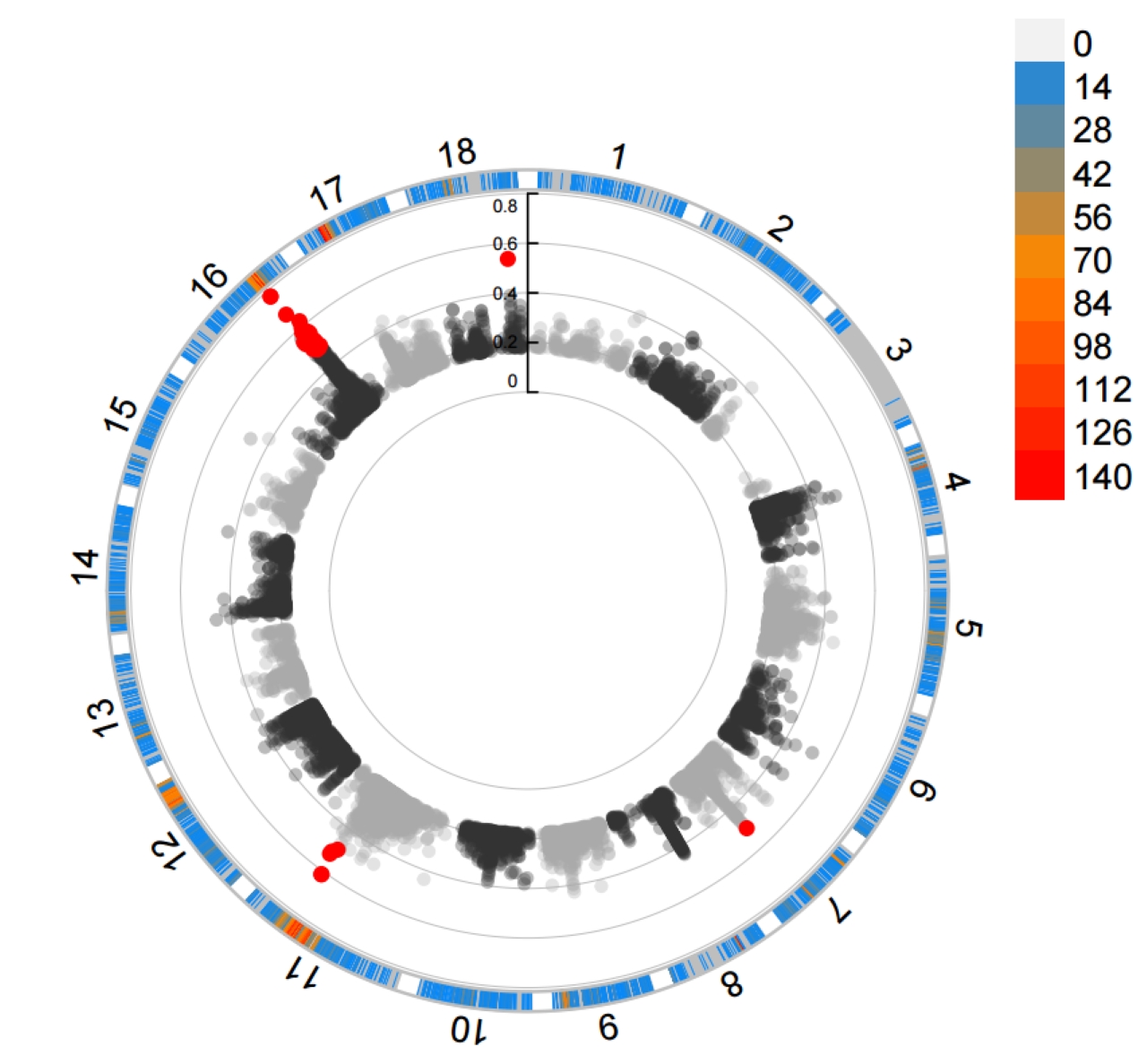
2.4.2 VCFtools FST results ./PopGeneticsVis/data/vcftools_fst/FST_JJ_G2_vs_HN_G2.windowed.weir.fst.chr.anno
head -n 10 ./PopGeneticsVis/data/vcftools_fst/FST_JJ_G2_vs_HN_G2.windowed.weir.fst.chr.anno
CHROM BIN_START BIN_END N_VARIANTS WEIGHTED_FST MEAN_FST anno_type gene_id
chr11 10001 20000 7 0.0436644 0.0239114 promoter,gene,UTR5,CDS,exon,intron,UTR3 HdF041849(p),HdF041849(g)
chr11 15001 25000 7 0.0436644 0.0239114 promoter,gene,UTR5,exon,intron HdF041849(p),HdF041849(g)
chr11 25001 35000 4 0.0150391 0.0161973 intergenic
chr11 30001 40000 4 0.0150391 0.0161973 intergenic
chr11 55001 65000 1 -0.00444874 -0.00444874 intergenic
chr11 60001 70000 1 -0.00444874 -0.00444874 intergenic
chr11 270001 280000 15 0.0511696 0.0366771 gene,UTR5,CDS,exon,intron,UTR3 HdF041844(g)
chr11 275001 285000 15 0.0511696 0.0366771 gene,UTR5,exon,intron HdF041844(g)
chr11 300001 310000 8 0.0526581 0.0448139 intergenic
Click to unfold the Table
| **CHROM** | **BIN_START** | **BIN_END** | **N_VARIANTS** | **WEIGHTED_FST** | **MEAN_FST** | **anno_type** | **gene_id** | |-----------|---------------|-------------|----------------|------------------|--------------|-----------------------------------------|---------------------------| | chr11 | 10001 | 20000 | 7.00E+00 | 0.0436644 | 0.0239114 | promoter,gene,UTR5,CDS,exon,intron,UTR3 | HdF041849(p),HdF041849(g) | | chr11 | 15001 | 25000 | 7.00E+00 | 0.0436644 | 0.0239114 | promoter,gene,UTR5,exon,intron | HdF041849(p),HdF041849(g) | | chr11 | 25001 | 35000 | 4.00E+00 | 0.0150391 | 0.0161973 | intergenic | | | chr11 | 30001 | 40000 | 4.00E+00 | 0.0150391 | 0.0161973 | intergenic | | | chr11 | 55001 | 65000 | 1.00E+00 | -0.00444874 | -0.00444874 | intergenic | | | chr11 | 60001 | 70000 | 1.00E+00 | -0.00444874 | -0.00444874 | intergenic | | | chr11 | 270001 | 280000 | 1.50E+01 | 0.0511696 | 0.0366771 | gene,UTR5,CDS,exon,intron,UTR3 | HdF041844(g) | | chr11 | 275001 | 285000 | 1.50E+01 | 0.0511696 | 0.0366771 | gene,UTR5,exon,intron | HdF041844(g) | | chr11 | 300001 | 310000 | 8.00E+00 | 0.0526581 | 0.0448139 | intergenic | |2.4.3 XPCLR XPCLR results ./PopGeneticsVis/data/xpclr_xpclr/JJ_G2_vs_HN_G2.xpclr.chr.anno
XPCLR (Cross-Population Composite Likelihood Ratio) is a commonly used analytical tool in population genetics for detecting selection signals, and it is particularly suitable for studying differences in genomic regions among different populations caused by natural selection. Its core principle is to calculate the “composite likelihood ratio” for each locus on the genome by comparing the differences in allele frequencies between two populations and incorporating linkage disequilibrium (LD) information, thereby identifying regions that may be affected by positive selection (or other selective pressures). Such regions usually exhibit population differentiation signals significantly higher than the genomic background level.
head -n 10 ./PopGeneticsVis/data/xpclr_xpclr/JJ_G2_vs_HN_G2.xpclr.chr.anno
chrom start stop pos_start pos_stop modelL nullL sel_coef nSNPs nSNPs_avail xpclr xpclr_norm anno_type gene_id
chr1 110001 120000 116131 119955 -35.0884425294439 -40.1089557348041 1e-04 16 16 10.0410264107203 -0.0638126714140551 intergenic
chr1 115001 125000 116131 124815 -66.45676465399 -66.885874161588 5e-05 26 26 0.858219015196113 -0.465779523577906 intergenic
chr1 120001 130000 121746 129981 -78.2152330276062 -88.4911108160588 1e-04 31 31 20.5517555769051 0.396282431847874 intergenic
chr1 125001 135000 125362 134705 -109.377249276282 -109.686346377196 1e-05 41 41 0.618194201828032 -0.476286334242127 intergenic
chr1 130001 140000 130307 139709 -108.695950408831 -115.431457733638 2e-04 50 50 13.4710146496142 0.0863311262244798 intergenic
chr1 135001 145000 135498 144985 -158.605704897143 -169.662861526333 4e-04 65 65 22.1143132583807 0.464681600587198 intergenic
chr1 140001 150000 141304 149997 -154.800677159078 -154.800677159078 0 54 54 0 -0.503347075759572 intergenic
chr1 145001 155000 145791 154842 -81.9384742813194 -82.1444387953709 5e-05 31 31 0.411929028102946 -0.485315355435985 intergenic
chr1 150001 160000 150028 159925 -44.0246980239117 -50.7171763729386 4e-04 19 19 13.3849566980539 0.0825640381867617 intergenic
Click to unfold the Table
| **chrom** | **start** | **stop** | **pos_start** | **pos_stop** | **modelL** | **nullL** | **sel_coef** | **nSNPs** | **nSNPs_avail** | **xpclr** | **xpclr_norm** | **anno_type** | |-----------|-----------|----------|---------------|--------------|--------------|--------------|--------------|-----------|-----------------|-------------|----------------|---------------| | chr1 | 110001 | 120000 | 1.16E+05 | 119955 | -35.08844253 | -40.10895573 | 1.00E-04 | 16 | 16 | 10.04102641 | -0.063812671 | intergenic | | chr1 | 115001 | 125000 | 1.16E+05 | 124815 | -66.45676465 | -66.88587416 | 5.00E-05 | 26 | 26 | 8.58E-01 | -0.465779524 | intergenic | | chr1 | 120001 | 130000 | 1.22E+05 | 129981 | -78.21523303 | -88.49111082 | 1.00E-04 | 31 | 31 | 20.55175558 | 0.396282432 | intergenic | | chr1 | 125001 | 135000 | 1.25E+05 | 134705 | -109.3772493 | -109.6863464 | 1.00E-05 | 41 | 41 | 0.618194202 | -0.476286334 | intergenic | | chr1 | 130001 | 140000 | 1.30E+05 | 139709 | -108.6959504 | -115.4314577 | 2.00E-04 | 50 | 50 | 13.47101465 | 0.086331126 | intergenic | | chr1 | 135001 | 145000 | 1.35E+05 | 144985 | -158.6057049 | -169.6628615 | 4.00E-04 | 65 | 65 | 22.11431326 | 0.464681601 | intergenic | | chr1 | 140001 | 150000 | 1.41E+05 | 149997 | -154.8006772 | -154.8006772 | 0 | 54 | 54 | 0 | -0.503347076 | intergenic | | chr1 | 145001 | 155000 | 1.46E+05 | 154842 | -81.93847428 | -82.1444388 | 5.00E-05 | 31 | 31 | 0.411929028 | -0.485315355 | intergenic | | chr1 | 150001 | 160000 | 1.50E+05 | 159925 | -44.02469802 | -50.71717637 | 4.00E-04 | 19 | 19 | 13.3849567 | 0.082564038 | intergenic |Rscript \
./PopGeneticsVis/bin/cmplot_fst_xpclr.R \
--method XPCLR \
--fst_xpclr ./PopGeneticsVis/data/xpclr_xpclr/JJ_G2_vs_HN_G2.xpclr.chr.anno \
--plot_type d
Rscript \
./PopGeneticsVis/bin/cmplot_fst_xpclr.R \
--method XPCLR \
--fst_xpclr ./PopGeneticsVis/data/xpclr_xpclr/JJ_G2_vs_HN_G2.xpclr.chr.anno \
--plot_type m
Rscript \
./PopGeneticsVis/bin/cmplot_fst_xpclr.R \
--method XPCLR \
--fst_xpclr ./PopGeneticsVis/data/xpclr_xpclr/JJ_G2_vs_HN_G2.xpclr.chr.anno \
--plot_type c
2.5 WGBS MethylDackel Cytosine Ratio Vis
cytosine_report (cytosine report) is a core file related to WGBS (Whole Genome Bisulfite Sequencing) data analysis. It is mainly used to store key information about cytosine sites in DNA methylation research, supporting subsequent methylation level visualization and Differential Methylation Region (DMR) analysis.
usage: ./PopGeneticsVis/bin/cytosine_ratio_vis.R [-h] --cytosine_report CYTOSINE_REPORT
[--sample_num SAMPLE_NUM]
Methyl Ratio Vis for SytosineReport
options:
-h, --help show this help message and exit
--cytosine_report CYTOSINE_REPORT
Cytosine report from Bismark/MethylDackel
[./cytosine_report.txt].
--sample_num SAMPLE_NUM
Methyl site number for visualization [1000000].
2.5.1 Terminal Running
Rscript \
./PopGeneticsVis/bin/cytosine_ratio_vis.R \
--cytosine_report ./PopGeneticsVis/data/cytosine_report/HN_G2_1.cytosine_report.txt \
--sample_n 1000000
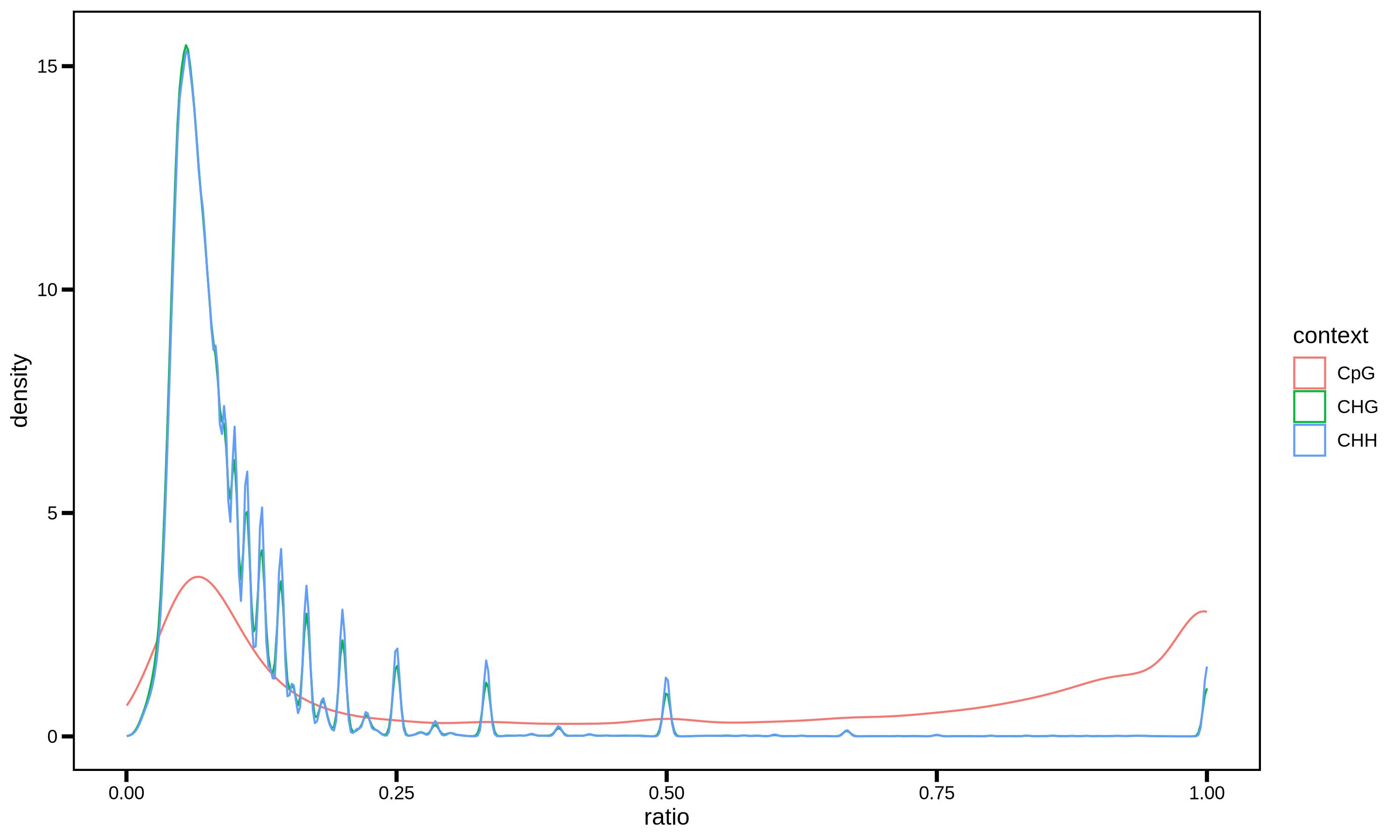
2.5.2 MethylDackel Cytosine Report ./PopGeneticsVis/data/cytosine_report/HN_G2_1.cytosine_report.txt
head -n 10 ./PopGeneticsVis/data/cytosine_report/HN_G2_1.cytosine_report.txt
mith1tg000589c 2 + 0 18 CHH CCC
mith1tg000589c 3 + 0 19 CHH CCC
mith1tg000589c 4 + 0 19 CHH CCC
mith1tg000589c 5 + 0 21 CHG CCG
mith1tg000589c 6 + 0 25 CG CGT
mith1tg000589c 7 - 0 0 CG CGG
mith1tg000589c 14 + 0 27 CG CGC
mith1tg000589c 15 - 0 0 CG CGT
mith1tg000589c 16 + 1 28 CHH CCC
mith1tg000589c 17 + 1 29 CHH CCT
Click to unfold the Table
| mith1tg000589c | 2 | + | 0 | 18 | CHH | CCC | |----------------|----|---|----------|----|-----|-----| | mith1tg000589c | 3 | + | 0.00E+00 | 19 | CHH | CCC | | mith1tg000589c | 4 | + | 0.00E+00 | 19 | CHH | CCC | | mith1tg000589c | 5 | + | 0.00E+00 | 21 | CHG | CCG | | mith1tg000589c | 6 | + | 0.00E+00 | 25 | CG | CGT | | mith1tg000589c | 7 | - | 0.00E+00 | 0 | CG | CGG | | mith1tg000589c | 14 | + | 0.00E+00 | 27 | CG | CGC | | mith1tg000589c | 15 | - | 0.00E+00 | 0 | CG | CGT | | mith1tg000589c | 16 | + | 1.00E+00 | 28 | CHH | CCC | | mith1tg000589c | 17 | + | 1.00E+00 | 29 | CHH | CCT |2.6 MethylKit DMR Analysis and Vis
usage: ./PopGeneticsVis/bin/methylkit_dmr.R [-h] --control CONTROL --treated TREATED --in_dir
IN_DIR --out_dir OUT_DIR [--threads THREADS]
[--nrep NREP] [--level {DMC,DMR,DMP}]
[--context {CpG,CHG,CHH}] [--mincov MINCOV]
[--win_size WIN_SIZE] [--step_size STEP_SIZE]
[--corr_method {pearson,kendall,spearman}]
[--adjust_method {SLIM,holm,hochberg,hommel,bonferroni,BH,BY,fdr,none,qvalue}]
[--test_method {F,Chisq,fast.fisher,midPval}]
[--qvalue_cutoff QVALUE_CUTOFF]
[--meth_cutoff METH_CUTOFF] [--gtf GTF]
[--promoter_up PROMOTER_UP]
[--promoter_down PROMOTER_DOWN]
[--plot_width PLOT_WIDTH]
[--plot_height PLOT_HEIGHT]
MethylKit Analysis Workflow
options:
-h, --help show this help message and exit
--control CONTROL Control group name.
--treated TREATED Treated group name.
--in_dir IN_DIR Input directory [./].
--out_dir OUT_DIR Output directory [./].
--threads THREADS Threads, more CPUs more Mems need [1].
--nrep NREP Samples replicates each group [3].
--level {DMC,DMR,DMP}
Methylation level [DMR].
--context {CpG,CHG,CHH}
Methylation context [CpG].
--mincov MINCOV Minimum coverage [10].
--win_size WIN_SIZE Window size for DMR (bp) [2000].
--step_size STEP_SIZE
Step size for DMR (bp) [2000].
--corr_method {pearson,kendall,spearman}
Correlation method [pearson].
--adjust_method {SLIM,holm,hochberg,hommel,bonferroni,BH,BY,fdr,none,qvalue}
P-value adjustment method [SLIM].
--test_method {F,Chisq,fast.fisher,midPval}
Statistical test method [Chisq].
--qvalue_cutoff QVALUE_CUTOFF
Q-value cutoff [0.05].
--meth_cutoff METH_CUTOFF
Methylation difference cutoff [25].
--gtf GTF GTF/GFF of genome [NULL].
--promoter_up PROMOTER_UP
Promoter upstream distance (bp) [2000]
--promoter_down PROMOTER_DOWN
Promoter downstream distance (bp) [200]
--plot_width PLOT_WIDTH
Plot width (inch) [10.00].
--plot_height PLOT_HEIGHT
Plot height (inch) [10.00].
2.6.1 Terminal Running
genome_gtf="/path_to_gtf/genome.gtf"
Rscript \
./PopGeneticsVis/bin/methylkit_dmr.R \
--control JJ_G2 \
--treated HN_G2 \
--in_dir ./PopGeneticsVis/data/cytosine_report/ \
--out_dir ./PopGeneticsVis/data/cytosine_report/ \
--threads 1 \
--nrep 3 \
--level DMR \
--context CpG \
--mincov 10 \
--win_size 2000 \
--step_size 2000 \
--corr_method pearson \
--adjust_method SLIM \
--test_method Chisq \
--qvalue_cutoff 0.05 \
--meth_cutoff 25 \
--gtf ${genome_gtf} \
--promoter_up 2000 \
--promoter_down 200 \
--plot_width 10.00 \
--plot_height 6.18
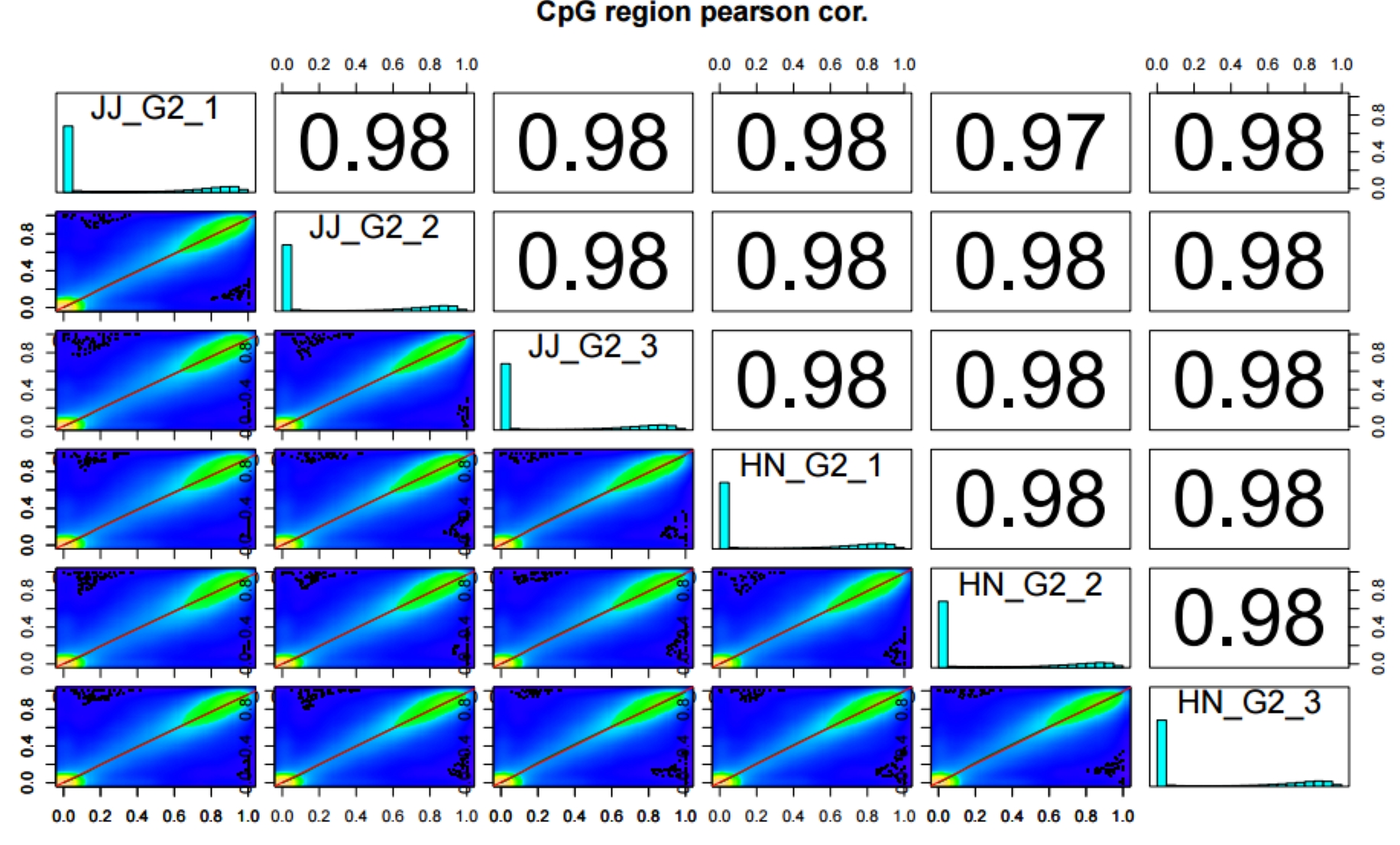
2.6.2 MethylDackel Cytosine Report ./PopGeneticsVis/data/cytosine_report/JJ_G2_1.cytosine_report.txt
head -n 10 ./PopGeneticsVis/data/cytosine_report/JJ_G2_1.cytosine_report.txt
chr2 1 + 0 0 CHG CTG
chr2 3 - 0 2 CHG CAG
chr2 5 + 0 0 CHH CAT
chr2 8 + 0 0 CHG CTG
chr2 10 - 2 1 CHG CAG
chr2 12 + 0 0 CHH CTC
chr2 14 + 0 0 CHH CCT
chr2 15 + 0 0 CHH CTC
chr2 17 + 0 0 CHH CTC
chr2 19 + 0 0 CHH CTC
Click to unfold the Table
| chr2 | 1 | + | 0 | 0 | CHG | CTG | |------|----|---|----------|---|-----|-----| | chr2 | 3 | - | 0.00E+00 | 2 | CHG | CAG | | chr2 | 5 | + | 0.00E+00 | 0 | CHH | CAT | | chr2 | 8 | + | 0.00E+00 | 0 | CHG | CTG | | chr2 | 10 | - | 2.00E+00 | 1 | CHG | CAG | | chr2 | 12 | + | 0.00E+00 | 0 | CHH | CTC | | chr2 | 14 | + | 0.00E+00 | 0 | CHH | CCT | | chr2 | 15 | + | 0.00E+00 | 0 | CHH | CTC | | chr2 | 17 | + | 0.00E+00 | 0 | CHH | CTC | | chr2 | 19 | + | 0.00E+00 | 0 | CHH | CTC |2.6.3 MethylKit DMR results ./PopGeneticsVis/data/cytosine_report/JJ_G2_vs_HN_G2_DMR_CpG_2k.txt
head -n 10 ./PopGeneticsVis/data/cytosine_report/JJ_G2_vs_HN_G2_DMR_CpG_2k.txt
chr start end strand pvalue qvalue meth.diff
chr1 94001 96000 * 0.359806000563359 0.387704326219981 0.152798303668958
chr1 96001 98000 * 0.470816921695121 0.456792909828252 -0.0848647129274156
chr1 98001 100000 * 0.000872125098979439 0.0030624412141788 -0.51115564900546
chr1 100001 102000 * 0.14767572092135 0.21438828041915 0.144018353504898
chr1 102001 104000 * 0.96580550253059 0.6773458835649 -0.00530964695794603
chr1 104001 106000 * 0.797705926039805 0.614400356560997 -0.0539165519981705
chr1 106001 108000 * 0.00835614419137723 0.0219464554487716 1.44288691348377
chr1 110001 112000 * 1.30699444113323e-06 8.11073044542023e-06 1.90364567269138
chr1 112001 114000 * 0.000328168930580941 0.00127959885901375 0.694565505097805
head -n 10 ./PopGeneticsVis/data/cytosine_report/JJ_G2_vs_HN_G2_DMR_CpG_2k_Sig.txt
chr start end strand pvalue qvalue meth.diff
chr1 466001 468000 * 0.00415711849409238 0.0120671167427604 -27.3170731707317
chr1 660001 662000 * 4.04113325784691e-09 3.62260475171775e-08 -33.3792540435737
chr1 1454001 1456000 * 8.93917792092073e-12 1.08386220427317e-10 -26.5563101301641
chr1 2750001 2752000 * 2.71610876608368e-129 5.71042756213395e-127 25.0474473214157
chr1 3428001 3430000 * 2.04907237864629e-10 2.14432834251086e-09 -46.3312368972746
chr1 3604001 3606000 * 2.13785433427291e-09 1.9841040640047e-08 37.8574305275876
chr1 4488001 4490000 * 3.46668153028929e-25 1.05232051309453e-23 -28.1260194808221
chr1 4934001 4936000 * 0.00170753601022128 0.00555652046196457 26.3565891472868
chr1 5294001 5296000 * 1.09733414650907e-08 9.28758216013033e-08 -58.5365853658537
Click to unfold the Table
| **chr** | **start** | **end** | **strand** | **pvalue** | **qvalue** | **meth.diff** | |---------|-----------|---------|------------|-------------|-------------|---------------| | chr1 | 94001 | 96000 | * | 0.359806001 | 0.387704326 | 0.152798304 | | chr1 | 96001 | 98000 | * | 0.470816922 | 0.45679291 | -0.084864713 | | chr1 | 98001 | 100000 | * | 0.000872125 | 0.003062441 | -0.511155649 | | chr1 | 100001 | 102000 | * | 0.147675721 | 0.21438828 | 0.144018354 | | chr1 | 102001 | 104000 | * | 0.965805503 | 0.677345884 | -0.005309647 | | chr1 | 104001 | 106000 | * | 0.797705926 | 0.614400357 | -0.053916552 | | chr1 | 106001 | 108000 | * | 0.008356144 | 0.021946455 | 1.442886913 | | chr1 | 110001 | 112000 | * | 1.31E-06 | 8.11E-06 | 1.903645673 | | chr1 | 112001 | 114000 | * | 0.000328169 | 0.001279599 | 0.694565505 |2.6.4 MethylKit DMR results ./PopGeneticsVis/data/cytosine_report/JJ_G2_vs_HN_G2_DMR_CpG_2k_Sig_Anno.txt
head -n 10 ./PopGeneticsVis/data/cytosine_report/JJ_G2_vs_HN_G2_DMR_CpG_2k_Sig_Anno.txt
chr start end strand pvalue qvalue meth.diff anno_genes anno_promoters
chr1 466001 468000 * 0.00415711849409238 0.0120671167427604 -27.3170731707317 HdF051575 NA
chr1 660001 662000 * 4.04113325784691e-09 3.62260475171775e-08 -33.3792540435737 HdF051575 NA
chr1 1454001 1456000 * 8.93917792092073e-12 1.08386220427317e-10 -26.5563101301641 HdF027216 NA
chr1 2750001 2752000 * 2.71610876608368e-129 5.71042756213395e-127 25.0474473214157 NA NA
chr1 3428001 3430000 * 2.04907237864629e-10 2.14432834251086e-09 -46.3312368972746 HdF027234 NA
chr1 3604001 3606000 * 2.13785433427291e-09 1.9841040640047e-08 37.8574305275876 HdF027242 NA
chr1 4488001 4490000 * 3.46668153028929e-25 1.05232051309453e-23 -28.1260194808221 HdF027261 NA
chr1 4934001 4936000 * 0.00170753601022128 0.00555652046196457 26.3565891472868 HdF027275 NA
chr1 5294001 5296000 * 1.09733414650907e-08 9.28758216013033e-08 -58.5365853658537 NA NA
Click to unfold the Table
| **chr** | **start** | **end** | **strand** | **pvalue** | **qvalue** | **meth.diff** | **anno_genes** | **anno_promoters** | |---------|-----------|---------|------------|-------------|-------------|---------------|----------------|--------------------| | chr1 | 466001 | 468000 | * | 0.004157118 | 0.012067117 | -27.31707317 | HdF051575 | NA | | chr1 | 660001 | 662000 | * | 4.04E-09 | 3.62E-08 | -33.37925404 | HdF051575 | NA | | chr1 | 1454001 | 1456000 | * | 8.94E-12 | 1.08E-10 | -26.55631013 | HdF027216 | NA | | chr1 | 2750001 | 2752000 | * | 2.72E-129 | 5.71E-127 | 25.04744732 | NA | NA | | chr1 | 3428001 | 3430000 | * | 2.05E-10 | 2.14E-09 | -46.3312369 | HdF027234 | NA | | chr1 | 3604001 | 3606000 | * | 2.14E-09 | 1.98E-08 | 37.85743053 | HdF027242 | NA | | chr1 | 4488001 | 4490000 | * | 3.47E-25 | 1.05E-23 | -28.12601948 | HdF027261 | NA | | chr1 | 4934001 | 4936000 | * | 1.71E-03 | 5.56E-03 | 26.35658915 | HdF027275 | NA | | chr1 | 5294001 | 5296000 | * | 1.10E-08 | 9.29E-08 | -58.53658537 | NA | NA |2.7 MethylKit Context Vis
MethylKit Context is a core analytical dimension in epigenetics (especially in DNA methylation analysis) based on the methylKit toolkit (commonly used for whole-genome bisulfite sequencing, WGBS, data analysis). It focuses on the distribution patterns and quantitative statistics of DNA methylation in different genomic contexts.
usage: ./PopGeneticsVis/bin/context_pie_vis.R [-h] --subjects_parts SUBJECTS_PARTS [--col_num COL_NUM]
Multi-Pie row: subjects; col: parts
optional arguments:
-h, --help show this help message and exit
--subjects_parts SUBJECTS_PARTS
Row: subjects; Col: parts [./subjects_parts.txt].
--col_num COL_NUM Columns/Pies in row [3].
2.7.1 Terminal Running
Rscript \
./PopGeneticsVis/bin/context_pie_vis.R \
--subjects_parts ./PopGeneticsVis/data/methyl_context/context_stats.txt \
--col_num 3
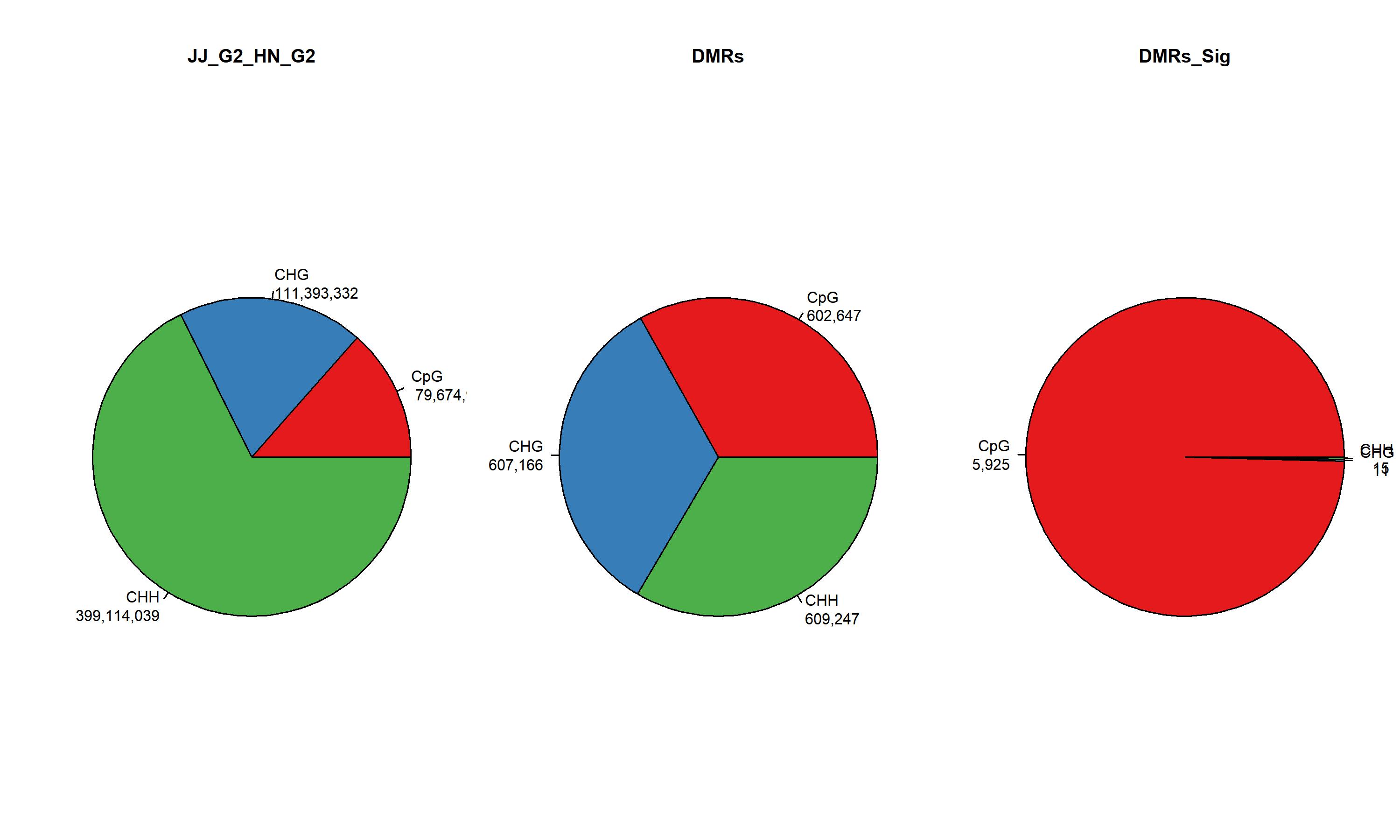
2.7.2 MethylKit Context Stats ./PopGeneticsVis/data/methyl_context/context_stats.txt
cat ./PopGeneticsVis/data/methyl_context/context_stats.txt
Subject CpG CHG CHH
JJ_G2_HN_G2 79674962 111393332 399114039
DMRs 602647 607166 609247
DMRs_Sig 5925 11 15
2.8 MethylKit DMR Violin Vis
MethylKit is an open-source bioinformatics toolkit based on the R language, specifically designed for processing high-throughput methylation sequencing data (such as WGBS, RRBS, MeDIP-seq, etc.) and analyzing differential methylation. Among these, Differentially Methylated Regions (DMRs) are one of its core analysis targets — that is, specific regions in the genome where there are significant differences in methylation levels between different sample groups (e.g., disease group vs. normal group, treatment group vs. control group). These regions are key entry points for 解析 the biological functions regulated by methylation (such as gene expression, cell differentiation, and disease occurrence).
usage: ./PopGeneticsVis/bin/methylkit_dmr_violin.R [-h] --methylkit_dmr METHYLKIT_DMR
[--violin_scale {area,count,width}]
[--violin_width VIOLIN_WIDTH]
[--point_size POINT_SIZE]
Meth.Diff Hyper & Hypo Violin Plot
options:
-h, --help show this help message and exit
--methylkit_dmr METHYLKIT_DMR
MethylKit DMR results (chr, start, end, ...,
meth.diff) [./methylkit_dmr.txt].
--violin_scale {area,count,width}
Violin area and width [count].
--violin_width VIOLIN_WIDTH
Violin width percent [1].
--point_size POINT_SIZE
Point size [1.5].
2.8.1 Terminal Running
Rscript \
./PopGeneticsVis/bin/methylkit_dmr_violin.R \
--methylkit_dmr ./PopGeneticsVis/data/methylkit_dmr/JJ_G2_vs_HN_G2_DMR_CpG_2k_Sig.txt \
--violin_scale count \
--violin_width 1 \
--point_size 1.5
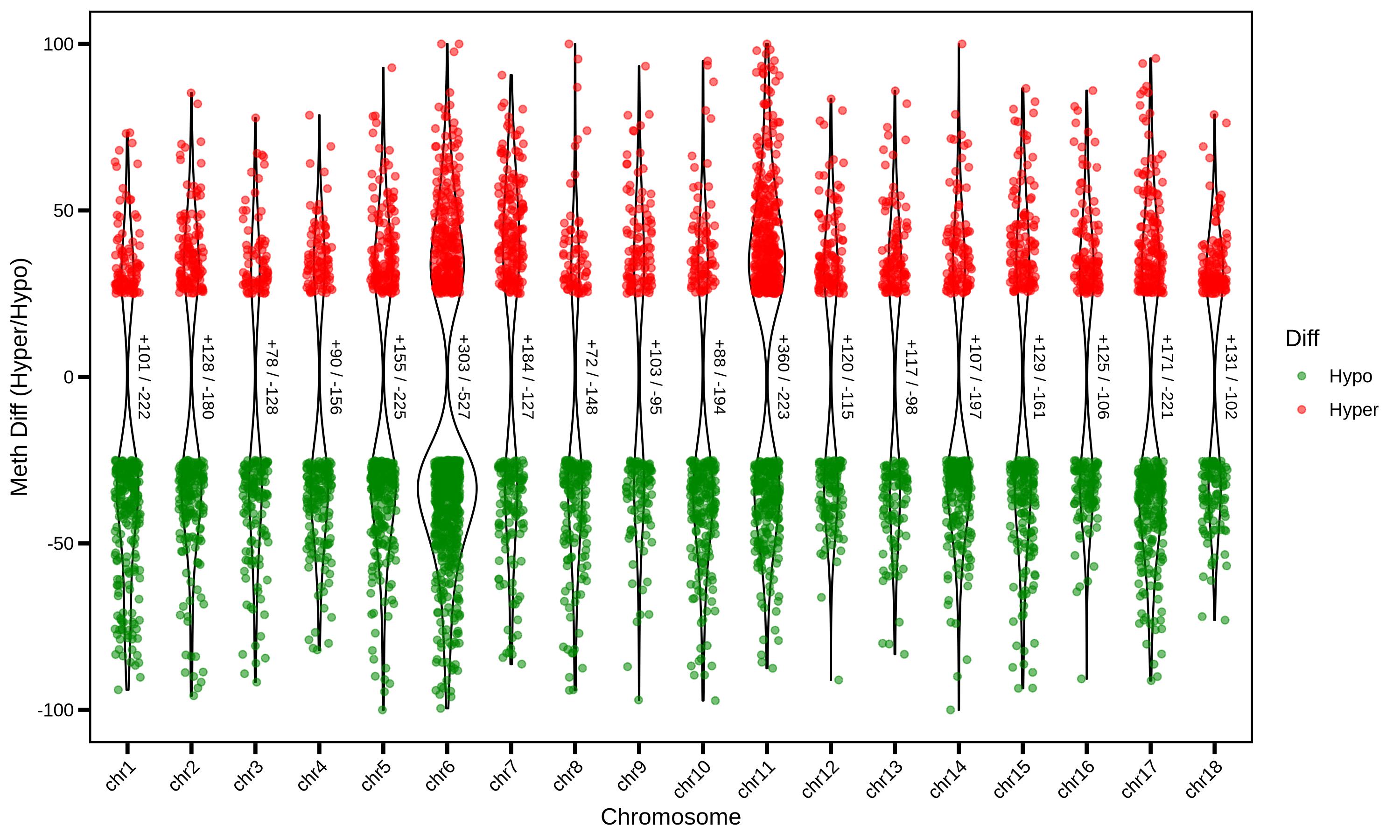
Rscript \
./PopGeneticsVis/bin/methylkit_dmr_violin.R \
--methylkit_dmr ./PopGeneticsVis/data/methylkit_dmr/JJ_G2_vs_HN_G2_DMR_CHG_2k_Sig.txt \
--violin_scale count \
--violin_width 1 \
--point_size 1.5
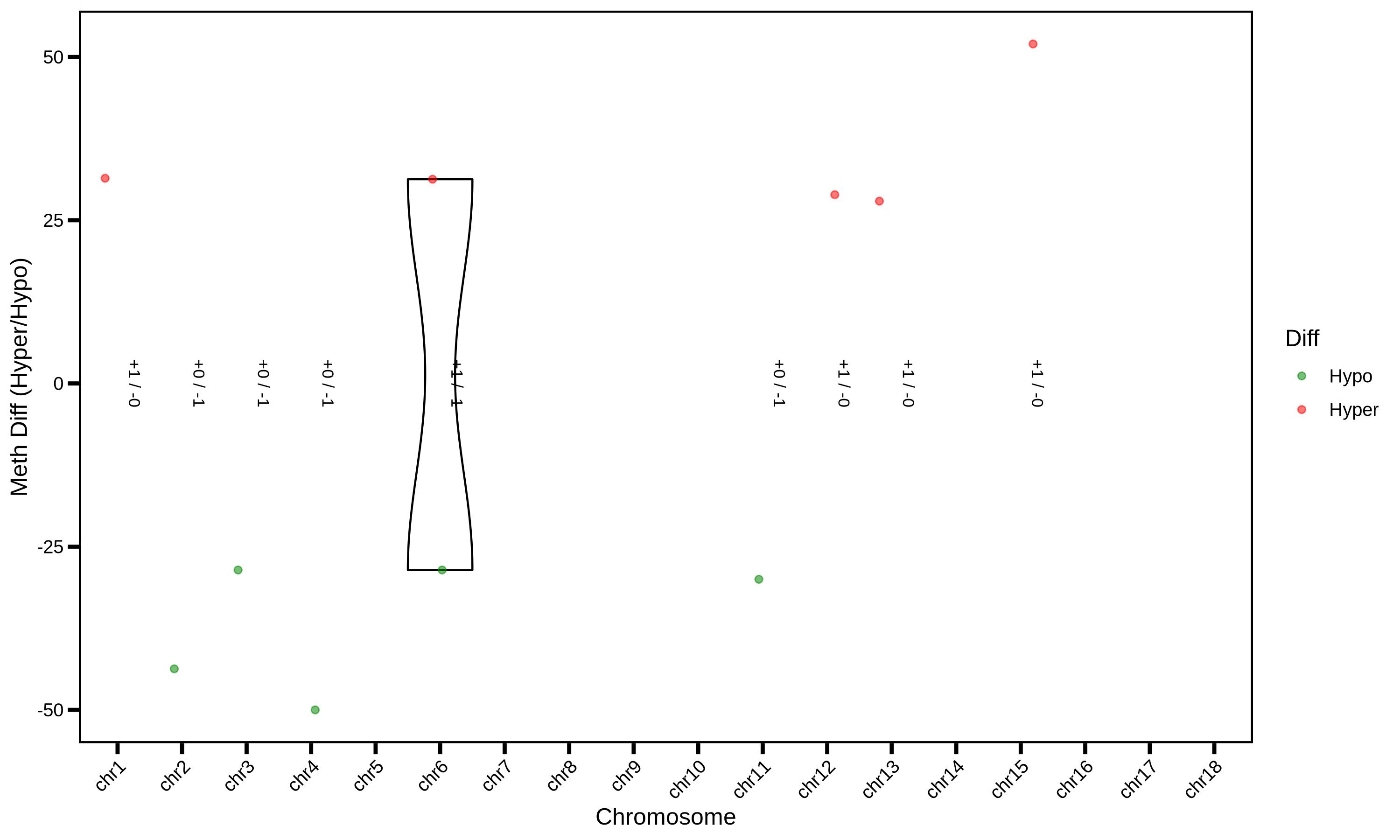
Rscript \
./PopGeneticsVis/bin/methylkit_dmr_violin.R \
--methylkit_dmr ./PopGeneticsVis/data/methylkit_dmr/JJ_G2_vs_HN_G2_DMR_CHH_2k_Sig.txt \
--violin_scale count \
--violin_width 1 \
--point_size 1.5
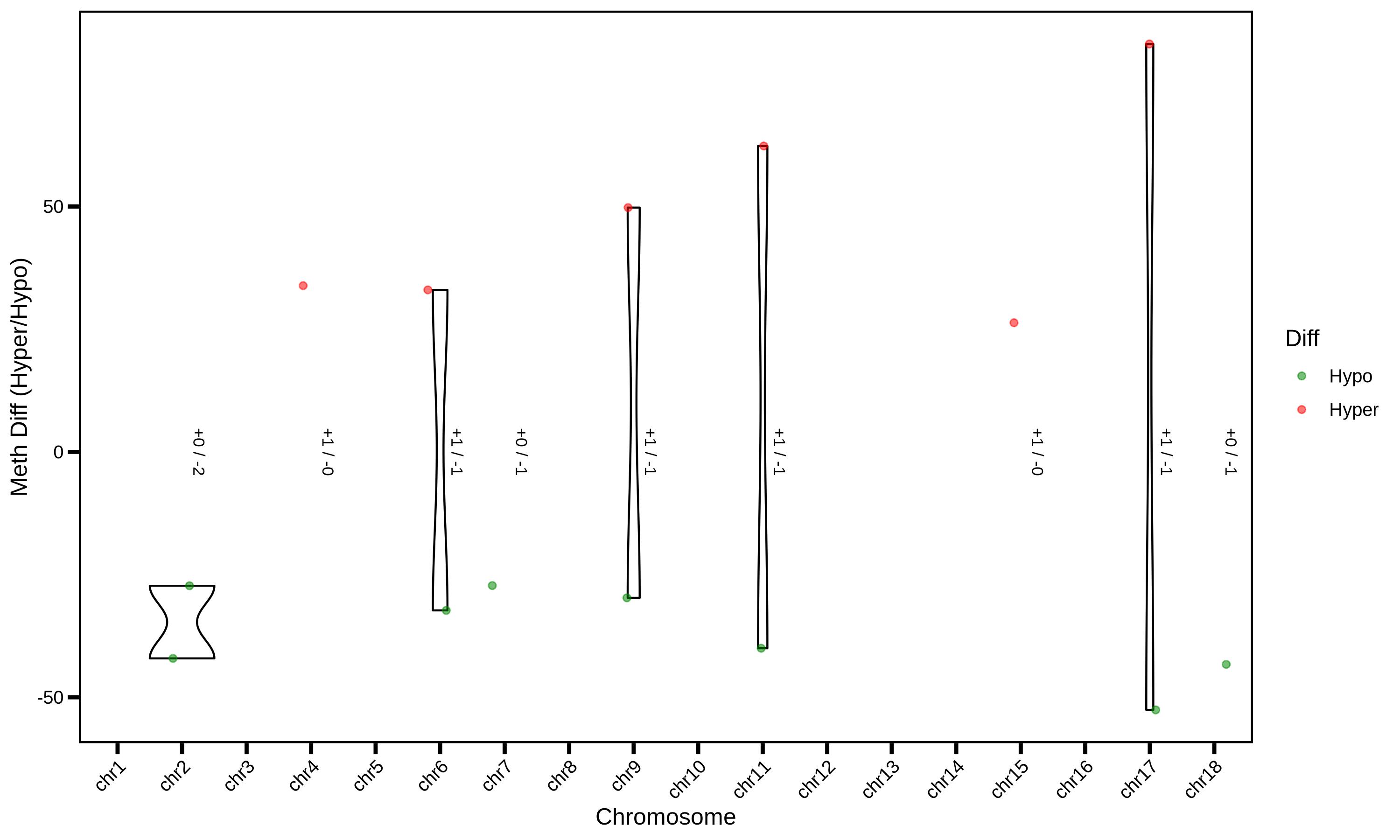
2.8.2 MethylKit DMR Sig ./PopGeneticsVis/data/methylkit_dmr/JJ_G2_vs_HN_G2_DMR_CpG_2k_Sig.txt
head -n 10 ./PopGeneticsVis/data/methylkit_dmr/JJ_G2_vs_HN_G2_DMR_CpG_2k_Sig.txt
chr start end strand pvalue qvalue meth.diff
chr1 466001 468000 * 0.00415711849409238 0.0120671167427604 -27.3170731707317
chr1 660001 662000 * 4.04113325784691e-09 3.62260475171775e-08 -33.3792540435737
chr1 1454001 1456000 * 8.93917792092073e-12 1.08386220427317e-10 -26.5563101301641
chr1 2750001 2752000 * 2.71610876608368e-129 5.71042756213395e-127 25.0474473214157
chr1 3428001 3430000 * 2.04907237864629e-10 2.14432834251086e-09 -46.3312368972746
chr1 3604001 3606000 * 2.13785433427291e-09 1.9841040640047e-08 37.8574305275876
chr1 4488001 4490000 * 3.46668153028929e-25 1.05232051309453e-23 -28.1260194808221
chr1 4934001 4936000 * 0.00170753601022128 0.00555652046196457 26.3565891472868
chr1 5294001 5296000 * 1.09733414650907e-08 9.28758216013033e-08 -58.5365853658537