Heatmap group for visualizing grouped gene expression data.
Source:R/heatmap_group.R
heatmap_group.RdHeatmap group for visualizing grouped gene expression data.
Usage
heatmap_group(
sample_gene,
group_sample,
scale_data = "row",
clust_method = "complete",
border_show = TRUE,
border_color = "#ffffff",
value_show = TRUE,
value_decimal = 2,
value_size = 5,
axis_size = 8,
cell_height = 10,
low_color = "#00880055",
mid_color = "#ffffff",
high_color = "#ff000055",
na_color = "#ff8800",
x_angle = 45
)Arguments
- sample_gene
Dataframe: Shared degs of all paired comparisons in all samples expression dataframe of RNA-Seq. (1st-col: Genes, 2nd-col~: Samples).
- group_sample
Dataframe: Samples and groups for gene expression (1st-col: Samples, 2nd-col: Groups).
- scale_data
Character: scale data. Default: "row", options: "row", "column", "none".
- clust_method
Character: cluster method. Default: "complete", options: "ward.D", "ward.D2", "single", "complete", "average" (= UPGMA), "mcquitty" (= WPGMA), "median" (= WPGMC) or "centroid" (= UPGMC).
- border_show
Logical: show border. Default: TRUE, options: TRUE, FALSE.
- border_color
Character: cell border color (color value or hex value with alpha). Default: "#ffffff".
- value_show
Logical: show value in cell. Default: TRUE, options: TRUE, FALSE.
- value_decimal
Numeric: cell value decimal. Default: 2, min: 0, max: 5.
- value_size
Numeric: cell value font size. Default: 5, min: 0, max: NULL.
- axis_size
Numeric: axis title font size. Default: 8, min: 0, max: NULL.
- cell_height
Numeric: cell height for value size and axis size. Default: 10.
- low_color
Character: min value color (color value or hex value with alpha). Default: "#00880055".
- mid_color
Character: min value color (color value or hex value with alpha). Default: "#ffffff".
- high_color
Character: min value color (color value or hex value with alpha). Default: "#ff000055".
- na_color
Character: min value color (color value or hex value with alpha). Default: "#ff8800".
- x_angle
Numeric: x axis text angle. Default: 45, min: 0, max: 360.
Examples
# 1. Library TOmicsVis package
library(TOmicsVis)
# 2. Use example dataset
data(gene_expression2)
head(gene_expression2)
#> Genes CT_1 CT_2 CT_3 LT20_1 LT20_2 LT20_3 LT15_1 LT15_2 LT15_3 LT12_1
#> 1 ACAA2 24.50 39.83 55.38 114.11 159.32 96.88 169.56 464.84 182.66 116.08
#> 2 ACAN 14.97 18.71 10.30 71.23 142.67 213.54 253.15 320.80 104.15 174.02
#> 3 ADH1 1.54 1.56 2.04 14.95 13.60 15.87 12.80 17.74 6.06 10.97
#> 4 AHSG 0.00 1911.99 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
#> 5 ALDH2 2.07 2.86 2.54 0.85 0.49 0.47 0.42 0.13 0.26 0.00
#> 6 AP1S3 6.62 14.59 9.30 24.90 33.94 23.19 24.00 36.08 27.40 24.06
#> LT12_2 LT12_3 LT12_6_1 LT12_6_2 LT12_6_3
#> 1 497.29 464.48 471.43 693.62 229.77
#> 2 305.81 469.48 1291.90 991.90 966.77
#> 3 10.71 30.95 9.84 10.91 7.28
#> 4 0.00 0.00 0.00 0.00 0.00
#> 5 0.28 0.11 0.37 0.15 0.11
#> 6 38.74 34.54 62.72 41.36 28.75
data(samples_groups)
head(samples_groups)
#> Samples Groups
#> 1 CT_1 CT
#> 2 CT_2 CT
#> 3 CT_3 CT
#> 4 LT20_1 LT20
#> 5 LT20_2 LT20
#> 6 LT20_3 LT20
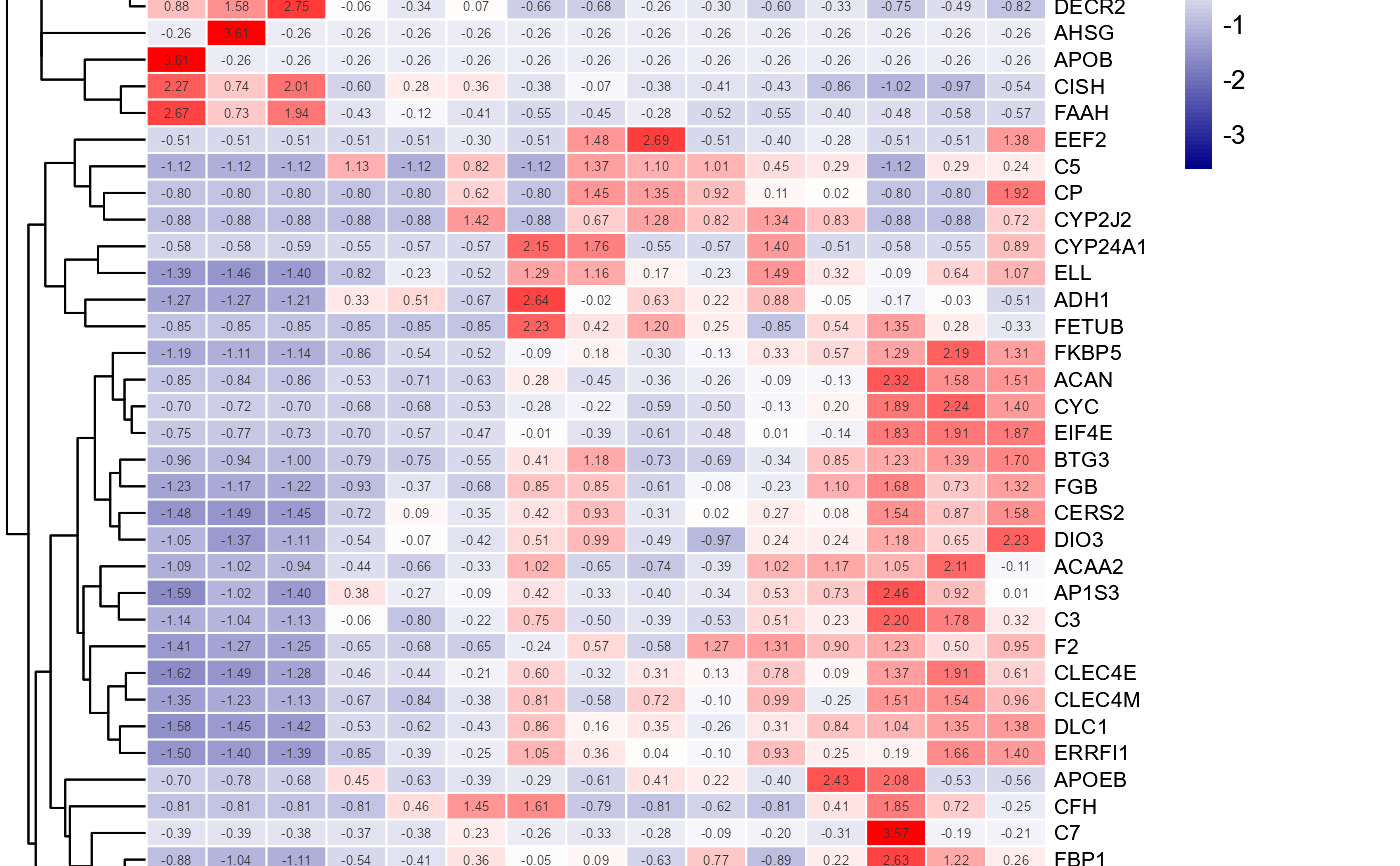
# 3. Default parameters
heatmap_group(gene_expression2[1:50,], samples_groups)
 # 4. Set scale_data = "column"
heatmap_group(gene_expression2[1:50,], samples_groups, scale_data = "column")
# 4. Set scale_data = "column"
heatmap_group(gene_expression2[1:50,], samples_groups, scale_data = "column")
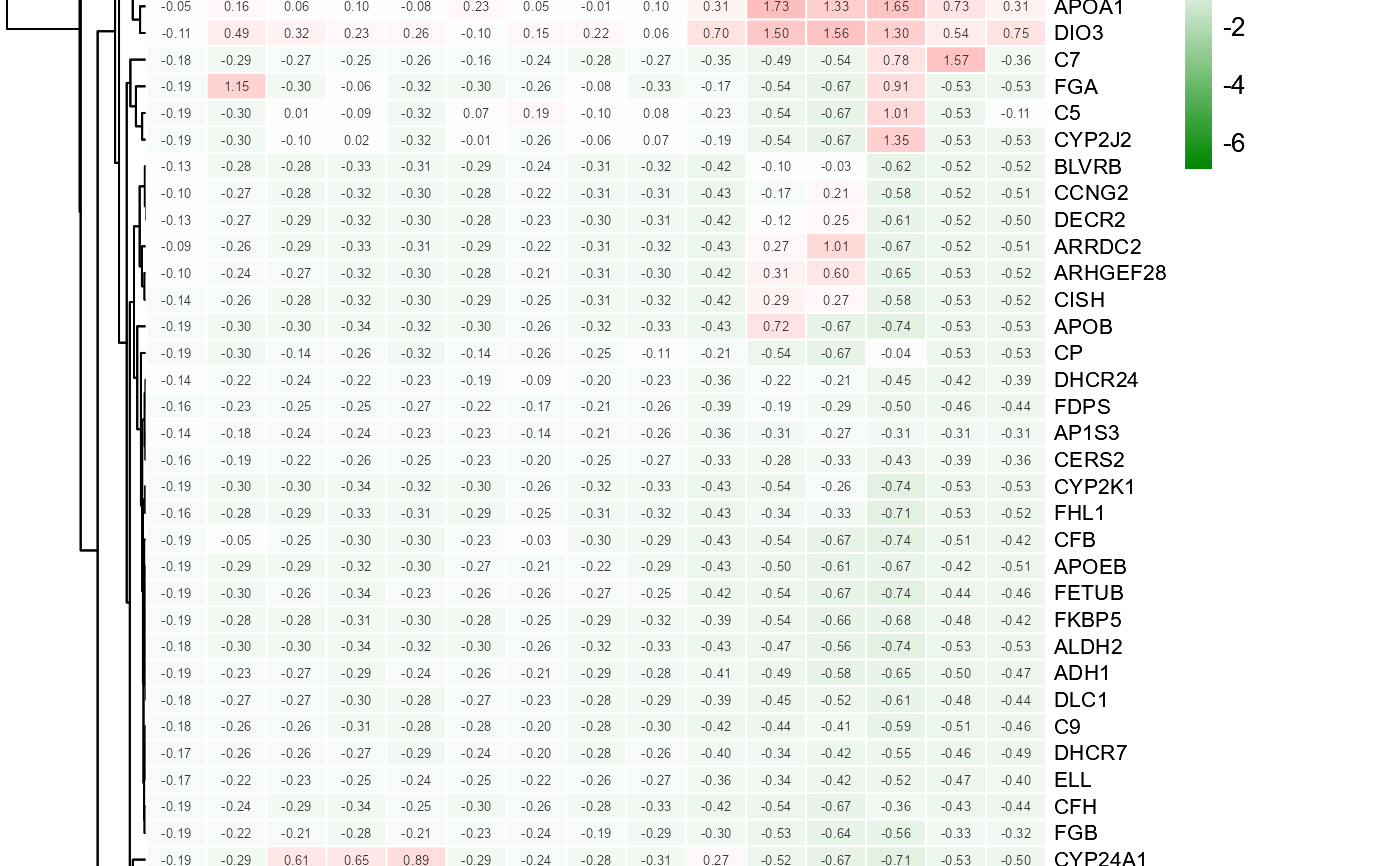 # 5. Set value_show = FALSE
heatmap_group(gene_expression2[1:50,], samples_groups, value_show = FALSE)
# 5. Set value_show = FALSE
heatmap_group(gene_expression2[1:50,], samples_groups, value_show = FALSE)
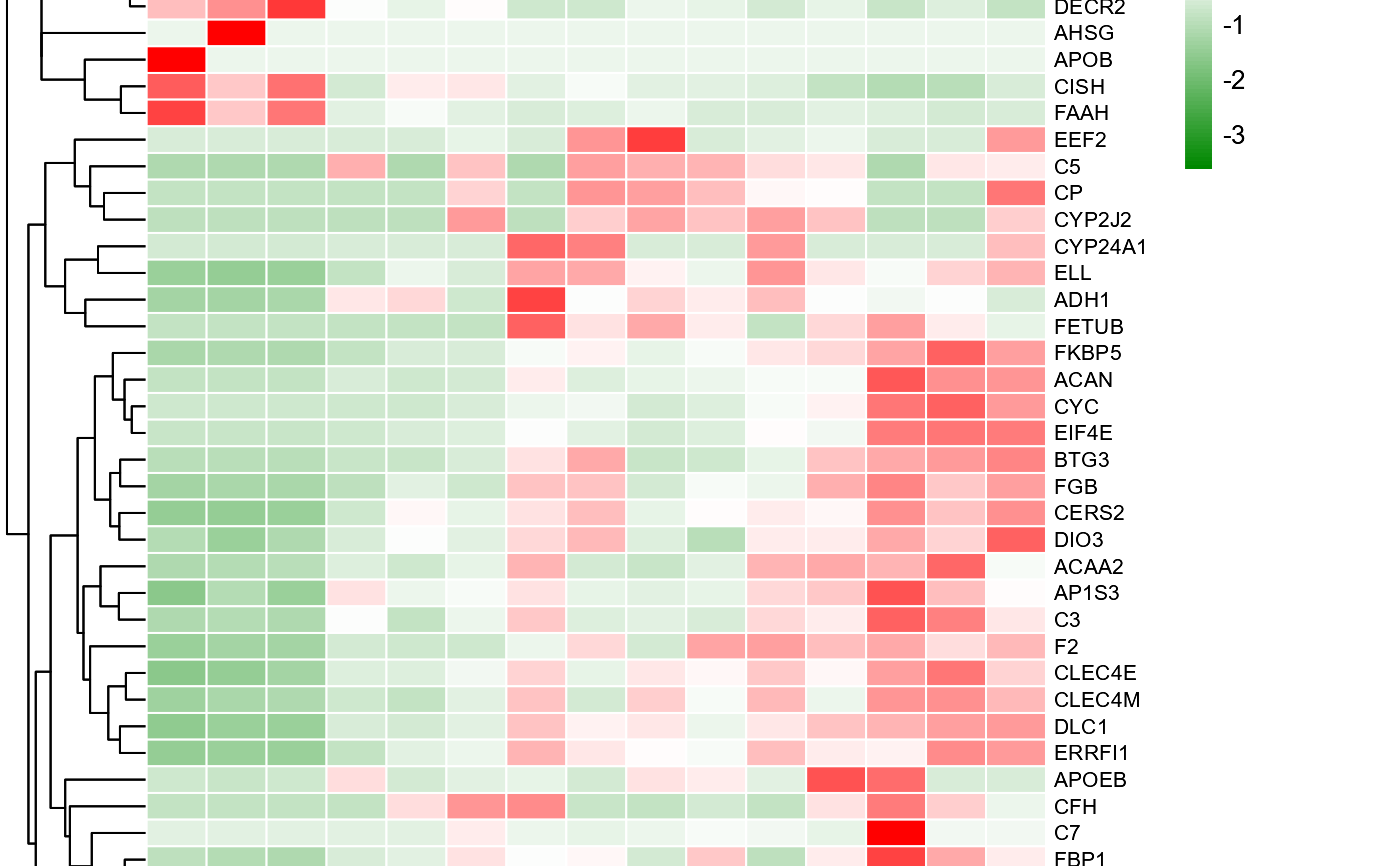 # 6. Set low_color = "#00008888"
heatmap_group(gene_expression2[1:50,], samples_groups, low_color = "#00008888")
# 6. Set low_color = "#00008888"
heatmap_group(gene_expression2[1:50,], samples_groups, low_color = "#00008888")